
Abstract
Chimeric antigen receptor (CAR) T-cell therapy is a revolutionary immunotherapy that has shown significant success in treating certain hematologic malignancies. However, there are still challenges and limitations associated with this therapy, and not all cancer patients benefit from this therapy alone. Therefore, modifying CAR-T-cell therapy based on immune checkpoints, and its combination with immune checkpoint inhibitors (ICIs), shows promise as a potentially more effective strategy for treating hematologic malignancies. This article outlines the progress of preclinical and clinical trials of CAR-T-cell therapy based on immune checkpoint modulation in the treatment of hematologic malignancies.
Checkpoint signaling interactions and pathways between tumor and immune cells.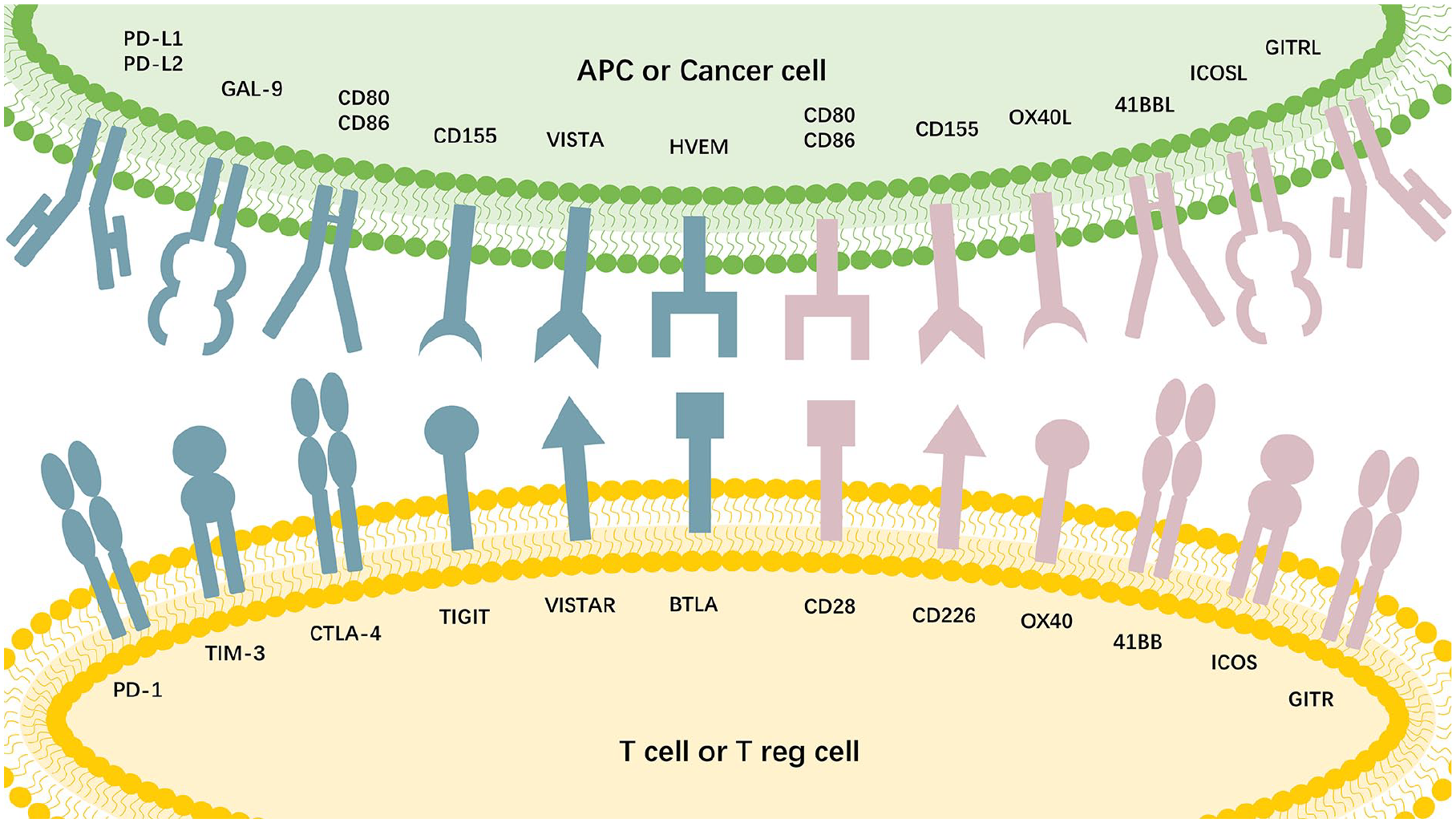
Introduction
Traditionally, the treatment of hematologic malignancies involved chemotherapy, radiotherapy, hematopoietic stem cell transplantation (HSCT), and targeted therapies. However, with continuous progress in cancer treatment, particularly the advent of chimeric antigen receptor (CAR) T-cell therapy, the landscape of treatment options for hematologic malignancy therapy has been revolutionized; this approach has achieved unprecedented success in recent years, especially in the treatment of B-cell acute lymphoblastic leukemia (B-ALL), with complete remission rates ranging from 70% to 90%1,2. Although CAR-T-cell therapy holds promise for novel therapeutic approaches in oncology, it may also trigger some serious adverse effects. Some patients may suffer from cytokine release syndrome (CRS) 3 , neurotoxicity 4 , and on-target off-tumor toxicity 5 , which can be life-threatening in extreme cases. In addition, although some patients will improve at the beginning of treatment, they may ultimately experience disease relapse factors such as genetic mutations, transient survival of CAR-T cells, immunogenicity of CAR-T cells themselves, tumor infiltration limits, related antigen escape, and CAR-T-cell depletion or immunosuppression microenvironment 6 . These challenges underscore the imperative to continue exploring alternative approaches to improve efficacy, mitigate side effects, and prolong the duration of therapy while advancing CAR-T-cell therapy. Therefore, seeking an approach to combine CAR-T-cell therapy with other treatment modalities is imperative.
Cancer cells tend to evade immune surveillance and resist destruction through various mechanisms. Immune evasion by cancer cells is of vital importance in the initiation, development, drug resistance, and poor prognosis of neoplasms. The mechanism underlying immunosuppression involves a multifaceted series of steps and factors, with the exhaustion of immune cells specialized in cell destruction, including CD8+ T cells and natural killer (NK) cells, playing a crucial role 7 . Tumors exploit immune evasion mechanisms to establish resilience to immune attacks, especially by suppressing the functionality of tumor-targeting T lymphocytes and NK cells 8 . The exhaustion of T/NK cells is typically characterized by increased expression of various immune checkpoint proteins (ICP), such as programmed cell death protein 1 (PD-1), cytotoxic T lymphocyte antigen-4 (CTLA-4), T-cell immunoglobulin and mucin domain-3 (TIM-3), lymphocyte activation gene-3 (LAG-3), B7 Homolog 3 (B7-H3), and T-cell activation V domain Ig inhibitor (VISTA). These ICPs act as “brakes,” inhibiting the anti-neoplastic capabilities of T cells and NK cells by interacting with specific ligands on antigen-presenting cells (APCs), cancer cells, and other constituents of the tumor microenvironment (TME)9,10. Interactions and pathways involving checkpoint signaling between tumor and immune cells are depicted in graphical abstract. Substantial evidence indicates that disrupting the interactiom between ICPs and their cognate ligands can reverse the immunocellular response against neoplastic growth, leading to tumor regression11,12.
Immune checkpoint inhibitors (ICIs) demonstrate therapeutic efficacy by reactivating exhausted immune responses, eliciting enduring clinical responses. Since the emergence of ICIs, there has been a significant enhancement in the overall survival (OS) rate of cancer patients, fundamentally altering the landscape of cancer treatment. ICIs have become the primary option for treating various cancers. Currently, numerous clinical trials for hematological malignancies are in progress 13 . Due to the varying levels of ICP expression and co-expression in different tumors, the therapeutic response to checkpoint inhibitors varies significantly. A meta-analysis indicates that approximately 80% of patients in clinical trials exhibited a lack of response to monotherapy utilizing PD-1/PD-L1 blockade, and some may even experience the detrimental phenomenon of excessive disease progression 14 . Cancers exhibiting responsiveness to ICIs are generally characterized by a significant T-cell infiltration within TME. In contrast, cancers that fail to elicit an endogenous T-cell response due to a plethora of factors, including the lack of a pronounced tumoral mutational load, demonstrate refractoriness to such immunotherapies. Consequently, a limitation of checkpoint therapy lies in the insufficient availability of T cells capable of responding to immune checkpoint therapy 15 . Furthermore, cancers that initially exhibit a response to checkpoint drugs may ultimately develop resistance through a range of genetic and immune-related mechanisms16,17. Hence, numerous clinical trials are presently assessing the impact of various ICIs when combined with other therapies for the management of hematological malignancies, such as radiotherapy, chemotherapy, and so on, which have been shown to enhance anti-tumor effects 18–20.
Therefore, combining CAR-T-cell therapy with ICI immunotherapy or modifying CAR-T-cell therapy based on immune checkpoints may be a more useful method for treating hematologic malignancies21,22. Recently, CAR-NK-cell therapy has been introduced as a supplement to CAR-T-cell therapy23,24. The consensus among most studies is that NK cells are considered safe in the allogeneic setting due to their lack of association with CRS, immune effector cell–associated neurotoxicity syndrome (ICANS), or graft-versus-host disease (GvHD) 25 . Moreover, CAR-NK cells can be mass-produced, thereby reducing the direct production costs 26 . CAR-NK-cell therapy faces various challenges that restrict its functionality and clinical application, which are comparable to those encountered by CAR-T-cell therapy 27 . The immune checkpoint, as a pivotal regulator of T-cell response, has garnered increasing attention in the realm of NK cells. Combining it with CAR-NK cells may potentially enhance therapeutic efficacy28,29. This article delivers the progress of preclinical and clinical trials of CAR-T-cell therapy based on immune checkpoint modulation in the treatment of hematologic tumors and briefly mentioned the relevant research on CAR-NK-cell therapy.
Factors Affecting the Efficacy of CAR-T Cells and TME
CAR-T-cell therapy has achieved remarkable progress in the treatment of hematological malignancies. However, it still encounters several challenges. Currently, factors influencing the efficacy of CAR-T-cell therapy are considered to include tumor burden, the persistence time of CAR-T cells, tumor antigen escape, immunosuppression in the TME, and so on. The TME is one of the extremely significant aspects. The TME is composed of non-cancer cells surrounding tumor cells and the molecules they secrete. The continuous interaction between tumor cells and the TME plays a decisive role in tumor genesis, progression, metastasis, and response to treatment 30 . Among them, cytokines play an important role, and they are the key factors of tumor immune regulation. Due to the presence of highly inhibitory cytokines, hypoxia, and reactive oxygen species, the TME is very unfavorable for the infiltration and effector function of T cells and CAR-T cells. The dynamic changes of cytokines play an important role in maintaining the immunosuppressive phenotype, and cytokines also play an important role in the development of suppression in TME, providing an opportunity to design new strategies and alternative options for TME immune regulation30,31. In addition, cytokines also play a multifaceted role in CAR-T-cell therapy. They are not only involved in the immune regulation of tumors but also affect the production process of CAR-T cells, including the use of cytokines in the expansion and differentiation of T cells. Overall, the role of cytokines in the TME is multifaceted 32 . To overcome these challenges, a large number of studies have concentrated on enhancing the function of CAR-T cells by modifying the TME33–35.
Programmed Cell Death Protein 1
PD-1 is an immunosuppressive molecule situated on the plasma membrane and a pivotal constituent of the B7/CD28 co-stimulatory molecule family. PD-1 is considered to be a checkpoint molecule involved in normal cell self-recognition during the immune surveillance response. PD-1 is highly expressed in B-cell malignancies, including B-cell lymphoma (BCL) 36 , non-Hodgkin lymphoma (NHL) 37 , mantle cell lymphoma (MCL) 38 , and acute myeloid leukemia (AML) 39 . High expression of this marker is conspicuously observed in macrophages, as well as in a subset of activated T and B lymphocytes and myeloid-derived suppressor cells (MDSCs) 40 . Blocking the PD-1 interaction is considered an important potential target for cancer therapy. Currently, various combinations of inhibitors that target the PD-1 signaling pathway and CAR-T cells are available. Anti-PD-1 monoclonal antibody (mAb) nivolumab and anti-PD-L1 mAb atezolizumab have been used in combination with CAR-T-cell therapy in some clinical trials, demonstrating certain efficacy and safety. A clinical study enrolled 44 patients with relapsed/refractory (R/R) diffuse large B-cell lymphoma (DLBCL). The experimental group underwent a combination therapy consisting of a PD-1 inhibitor along with anti-CD19 CAR-T-cell therapy, while the remaining patients as the control group received only anti-CD19 CAR-T-cell therapy. The findings indicated that the objective response rate (ORR) for the experimental group was 65.39%, and for the control group, it was 61.11%, with no statistically significant difference (P > 0.05). The progression-free survival (PFS) and OS at 3 and 12 months after CAR-T-cell infusion were greater in the combination therapy group than those in the control group. The severity of CRS and ICANS was similar between the two groups. In the sustained treatment group, merely a few cases experienced grade 1 or grade 2 adverse events (CTCAEs), and no patients discontinued maintenance therapy as a result. This study suggests that although combination therapy did not improve the ORR, patients who achieved ORR after combination therapy may derive advantage from PD-1 inhibitor maintenance therapy without experiencing augmented adverse effects. This study indicates that sustained PD-1 inhibitor therapy may be a viable treatment option for R/R DLBCL patients with a high tumor burden, but additional extensive research and prolonged monitoring are required to validate these results 41 . Researchers developed CAR-T cells that secrete PD-1 blocking single-chain variable fragments (scFvs) through a suite of in vivo and in vitro experiments. These CAR-T cells enhance anti-tumor activity and the therapeutic potential of CAR-T cells via both autocrine and paracrine mechanisms. In a study by Rafiq et al., T cells were genetically modified to secrete a targeted anti-PD-1–blocking scFv in conjunction with PD-L1-positive Nalm6. In comparison with traditional CAR-T cells, CAR-T cells modified with self-secreted scFv PD-L1 antibodies exhibited increased cytotoxicity. As the secreted scFv is targeted at the tumor region, it protects CAR-T cells from PD-1 inhibition, potentially mitigating toxicities stemming from systemic checkpoint inhibition, thereby improving safety. This approach more effectively suppresses cancer and significantly prolongs the survival of mice. The article suggested that by harnessing scFvs that target alternative molecules, this immunomodulatory CAR-T-cell–dependent approach can be expanded and may potentially augment the therapeutic outcomes of CAR-T-cell and ICI-based therapies 42 . In a phase 1 clinical study, a total of nine patients received infusions of a novel dominant-negative PD-1-expressing anti-CD19 CAR-T cell at doses exceeding 1 × 106/kg. The ORR was 77.8%, with a CR rate of 55.6%. Two patients maintained CR for more than 20 months. Of all patients, 11.1% exhibited grade 3 or higher CRS and neurotoxicity. CAR-T cells proliferate following infusion and remained detectable for more than 12 months in patients who achieved CR. This article indicates that this novel CD19 CAR-T-cell therapy is safe and effective for treating patients with R/R DLBCL and promotes durable remission. The potential of this approach has been preliminarily validated in clinical research, providing a promising new approach for the treatment of R/R DLBCL. Further studies and protracted monitoring are needed to fully assess its advantages and risks and to determine its exact place in clinical practice 43 . In a phase Ib study, the research team prepared a novel CD19-specific CAR-T cell expressing a PD-1/CD28 chimeric switch receptor (CD19-PD-1/CD28-CAR). Researchers have demonstrated that it can enhance the proliferation of T cells and their cytotoxicity against BCL cells expressing PD-L1 in vitro and in vivo experiments. A first-in-human study was conducted, in which 10 out of 17 patients with R/R lymphoma achieved objective responses (58.8%). After a median duration of 15 months after follow-up, the median OS for the entire patient cohort remained undetermined. Significantly, there were no instances of severe neurotoxicity or CRS, which indicates promising therapeutic effectiveness 44 . The center also reported that six patients with R/R DLBCL who progressed after CD19 CAR-T-cell therapy were treated with CD19-specific CAR-T cells expressing CD19-PD-1/CD28 CAR-T cells as a last-resort treatment. Three of these patients achieved CR, with response durations ranging from 8 to 25 months. One patient experienced disease stabilization, and two patients died within 2 months due to disease advancement. Significantly, no instances of severe neurotoxicity or CRS were detected. These findings indicate that CD19-PD-1/CD28 CAR-T cells are a novel anti-CD19 CAR-T-cell therapy that can generate effective and lasting anti-tumor responses and can serve as a therapeutic option after the ineffectiveness of CD19 CAR-T-cell therapy 45 . The prognosis for patients with R/R AML is unfavorable and often exhibits resistance to chemotherapy, necessitating the urgent development of novel treatment strategies. One study reported the efficacious utilization of PD-1-silenced CAR-T-cell therapy targeting C-type lectin-like molecule-1 (CLL-1) in two R/R AML patients who were unresponsive to multiple salvage therapies. CLL-1 exhibits robust expression on AML cells, contrasting with its negligible presence on standard hematopoietic stem cells (HSCs), thereby rendering it a prime candidate for targeted immunotherapeutic approaches in AML patients. Genetically modified CAR-T cells can recognize and bind to CLL-1 molecules on AML cells in vivo, activating T cells and directing them to eliminate target cells. In addition, through PD-1-silencing, CAR-T cells can resist immunosuppressive signals in TME, maintaining their activity and proliferative capacity and thereby eradicating leukemia cells more effectively 46 . Both patients in the study received PD-1-silenced anti-CLL-1 CAR-T-cell therapy and achieved CR at 28 days after treatment, although the hematologic recovery was incomplete. The patients experienced CRS classified as grades 1 and 2. At the final follow-up, both patients achieved response (CR) for periods of 8 and 3 months. This article emphasizes the secure and potent nature of PD-1-silenced CLL-1 CAR-T-cell therapy in adult AML patients. However, due to the limited generalizability of the results from only two specific cases, the findings may not be applicable to all R/R AML patients. As a case report, the study design itself does not provide evidence of causality, and further clinical trials and randomized controlled trials are essential for substantiating the therapeutic efficacy 47 . In addition, CAR-T cells targeting Epidermal Growth Factor Receptor variant III (EGFRvIII) with PD-1 knockout have demonstrated enhanced anti-tumor activity in preclinical trials against solid tumors, such as glioma 48 . A substantial body of evidence indicates that blockade of the PD-1/PD-L1 pathway can directly modulate the function of NK cells in mice and humans49–51. Researchers led by Fabian have reported for the first time on a novel NK cell line, known as PD-L1 targeting high-affinity NK (t-haNK) cells, which have been genetically engineered to express high-affinity CD16, IL-2 retained in the endoplasmic reticulum, and a CAR for PD-L1. The study has confirmed that these PD-L1 t-haNK cells possess significant anti-tumor capabilities 52 . Liu et al. 53 demonstrated that dual therapy with CAR-NK cells and nivolumab produces a synergistic anti-tumor response in a humanized mouse cancer model. The clinical trials of the PD-1 in combination with CAR-T-cell therapy are delineated in Table 1.
The Clinical Trials of the PD-1 in Combination With CAR-T-Cell Therapy.
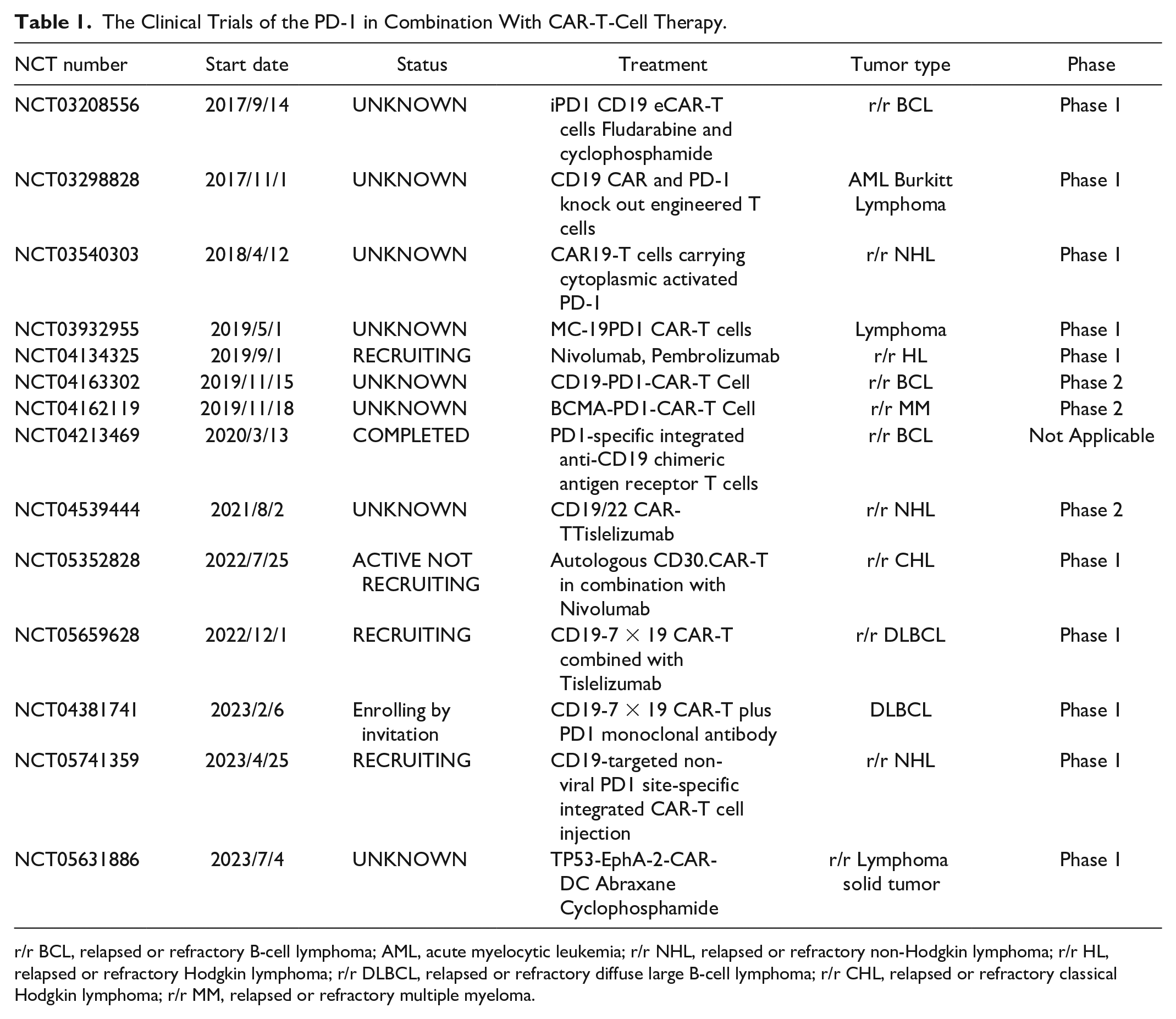
r/r BCL, relapsed or refractory B-cell lymphoma; AML, acute myelocytic leukemia; r/r NHL, relapsed or refractory non-Hodgkin lymphoma; r/r HL, relapsed or refractory Hodgkin lymphoma; r/r DLBCL, relapsed or refractory diffuse large B-cell lymphoma; r/r CHL, relapsed or refractory classical Hodgkin lymphoma; r/r MM, relapsed or refractory multiple myeloma.
Cytotoxic T-Lymphocyte Antigen-4
CTLA-4 is a surface protein that is a homolog of CD28 and is mainly involved in the immune evasion of malignant transformed cells. When CD28 binds to overexpressed CTLA-4, it blocks the co-stimulatory effect and T-cell priming 54 . Upon costimulation via the T-cell receptor (TCR) and the B7-CD28 co-stimulatory pathway, the expression of this protein is elevated, facilitating the attenuation of immune responses and averting autoimmunity. Anti-CTLA-4 impede the inhibitory signals, thereby stimulating the activation of effector T cells, and they also diminish the presence of tumor-infiltrating regulatory T cells (Tregs) via mechanisms that include antibody-mediated cellular cytotoxicity (ADCC) and antibody-mediated phagocytosis (ADCP) 55 . Currently, the immunosuppressive agents developed against CTLA-4 are ipilimumab and tremelimumab, which are primarily utilized in the treatment of solid tumors56–58. One study engineered a novel CAR by fusing the extracellular and transmembrane regions of CTLA-4 with the signaling domains of CD28 and the CD3zeta chain, CTLA-4-CD28-CAR-T cells. This method can enhance the specificity and potency of the immune response against tumors by targeting cancer cells that express CD80/CD86. The CTLA-4 CAR-T cells exhibited potent cytotoxicity to CD80/CD86-positive tumor cells, while sparing the CD80/CD86-negative K562 cells. Moreover, CTLA-4 CAR-T cells mitigated the proliferation of xenografted malignancies, particularly in the context of MDSCs, and did so without inducing CRS 59 . CAR function can be reprogrammed by fusing CTLA-4 cytoplasmic tails (CCTs) with monomeric, double-stranded, or triple-stranded CTLA-4 to the C-terminus of CAR, leading to a steady augmentation of cytotoxic potency accompanied by a decrease in proinflammatory cytokine activation and production. The fusion CAR has greater persistence and superior anticancer efficacy in models of relapsed leukemia. Single-cell RNA sequencing and flow cytometry analysis revealed that CAR2CCT cells retained a more pronounced central memory T-cell phenotype and exhibited greater persistence 60 . Presently, investigations are underway to determine if the inactivation of co-inhibitory receptors such as CTLA-4 or PD-1 can rejuvenate the potency of CAR-T-cell therapies. Using a humanized CD19-specific scFv coupled with a CD8a hinge and transmembrane segment, along with a fusion of the CD137 (4-1BB) and CD3z intracellular signaling domains, CD19 CAR-T cells were engineered from wild-type (WT) and normal donor (ND) T cells that were devoid of PDCD1 or CTLA-4. Crispr-Cas9-mediated deletion of PDCD1 or CTLA-4 exhibited the ability to potentiate the anticancer efficacy of CD19 CAR-T cells in in vitro and in vivo models of CAR-T cells dysfunction. The results demonstrated that CTLA-4-deficient CD19 CAR-T cells proved better expansion and better anticancer efficacy than PD-1-deficient CD19 CAR-T cells and CD19 CAR-T cells lacking both PD-1 and CTLA-4. This study suggested that CTLA-4 deletion allows uninhibited CD28 signaling and maintains T-cell surface CAR expression with a high concentration of antigen, thereby promoting homologous antigen-specific cytotoxicity. Therefore, deletion of CTLA-4 by CRISPR/Cas9 could enhance CD28 signaling and restore the adaptive and anti-tumor functions of CAR-T cells, including those derived from patients experiencing treatment failure with CAR-T-cell therapy 61 . Although this study provides important evidence that CTLA-4 loss enhances CAR-T-cell function, these findings may vary in different biological contexts, such as depending on the tumor type, antigen expression level, and expression of other immune checkpoint molecules. In the past, CTLA-4 was thought to be expressed solely on T cells, but recent findings have revealed its expression on tumors and NK cells as well 62 . Anti-CTLA-4 mAbs may indirectly enhance the activity of NK cells by inducing the secretion of cytokines from effector T cells and/or by depleting Tregs 28 . This could represent a significant breakthrough for improving the efficacy of CAR-NK-cell therapy in the future.
T-Cell Immunoglobulin and Mucin Domain-3
Up to now, expression of TIM-3 has been observed on a spectrum of immune cells such as T cells excluding the Th2 subset, NK cells, macrophages, dendritic cells (DCs), MDSCs, and mast cells. In addition, TIM-3 expression is also observed on specific cancerous cells, including melanoma and leukemia cells, and mainly binds to galectin-9 on APCs and triggers the apoptosis of T cells, thereby facilitating tumor cell growth and persistence7,28,63. Thus far, over 30 mAbs targeting TIM-3 have been assessed in clinical trials for cancer immunotherapy. In addition, TIM-3 expression is prevalent in the majority of leukemia stem cells (LSCs) in AML, contrasting with its absence in normal HSCs, highlighting its potential as a therapeutic target64,65. Chiba et al. 66 developed a second-generation CD19 CAR-T-cell therapy that integrates a 41BB co-stimulatory domain linked to a T3/28 chimeric protein. The utilization of the T3/28 chimeric protein notably extended the longevity of CAR-T cells in vivo. In addition, the robust anti-tumor efficacy demonstrated in mouse models of B lymphoma has shed new light on strategies for adoptive T-cell therapy 66 . Similarly, Zhao et al. 67 engineered CAR-T cells with a TIM-3 focus, enhancing their therapeutic longevity and exhibiting strong anti-tumor capabilities in vitro and within a murine model of multiple myeloma (MM). Lee et al. 68 used a naive human Fab phage display library to isolate anti-human TIM-3 antibodies and design an advanced generation of anti-TIM-3 antibodies. Treatment with anti-tim-3 CAR-T cells led to a substantial decrement in the neoplastic load in the hepatic and hematopoietic compartments of mice at the 2- to 3-week follow-up period after therapy. In vitro cytotoxicity experiments also showed that the anti-TIM-3 CAR-T cells could recognize and eliminate patient-isolated TIM-3+CD34+ LSCs, with the TIM-3-CD34+ T cells being conserved, indicating that these cells have antigen-directed potential to kill resistant LSCs. In conclusion, anti-TIM-3 CAR-T cells exhibited strong anti-myeloid leukemia efficacy both ex vivo and within a murine model of AML. More importantly, researchers demonstrated the potent cytotoxic effect on native LSCs derived from patients. Therefore, employing anti-TIM-3 CAR-T-cell therapy after the initial treatment to target LSCs in minimal residual disease (MRD) could potentially enhance the clinical outcomes for AML patients 68 . The team led by Blaeschke has engineered an array of TIM-3-CD28 chimeric proteins that transform the suppressive signal of TIM-3 into a stimulatory signal for T cells. In addition to anti-CD19 CAR-T cells, the two CD28-enriched chimeric receptor variants also exhibited better cytokine production and cell proliferation upon stimulation with CD3 antibody. Integration of these innovative chimeric proteins with both first- and second-generation anti-CD19 CAR-T cells resulted in a pronounced enhancement of their proliferative, activating, and cytotoxic capabilities. Their research suggests that the incorporation of checkpoint fusion proteins with anti-CD19 CAR-T cells can notably amplify T-cell expansion and surmount the suppressive signals mediated by the extracellular domain of TIM-3, particularly when integrated with second-generation CAR-T cells, leading to heightened therapeutic efficacy and longevity. Looking ahead, they intend to further investigate the potential of these TIM-3-CD28 chimeric proteins through a series of assays, including interactions with various CAR specificities, inhibition studies, and co-incubation experiments with native B-precursor cells. Moreover, they aim to evaluate their therapeutic potential in pertinent in vivo models, such as traditional xenograft or patient-derived xenograft models, to explore their additional benefits in the context of oncology 69 . Although TIM-3 is highly expressed in certain hematologic malignancies, it is also expressed in normal tissues, which may increase the risk of nonspecific toxicity during treatment. Therefore, more precise targeting strategies need to be developed to ensure treatment safety. At present, CAR-T-cell therapy targeting TIM-3 is still in the early research phase, and clinical data are relatively limited. More clinical trials are imperative for validating the effectiveness and safety profile of this treatment approach and to determine the optimal treatment strategy.
Lymphocyte Activation Gene-3
LAG-3, a transmembrane protein, serves as a crucial inhibitory receptor, exhibiting a structural resemblance to CD4 and consequently occupies an important position in immune regulation. Under physiological conditions, LAG-3 is predominantly expressed on activated T cells, B cells, NK cells, and DCs, exerting a negative regulatory effect on T-cell function 70 . It has also been identified as an expressed marker on a distinct subpopulation of malignant B cells in individuals diagnosed with DLBCL 71 . Accumulating evidence suggests that LAG-3 negatively affects the effector capability of T cells both in vivo and in vitro and also augments the inhibitory function of Treg cells. This indicates a crucial significance of LAG-3 in regulating the balance between effector and regulatory immune responses72–74. In a study by Zhang et al. 2 , CRISPRCas9 was used to knockout LAG-3 expression on human T cells and CAR-T cells; LAG-3 knockout CAR-T cells exhibited strong and specific anti-tumor activity mediated by antigens with enhanced T-cell expansion and cytokine production in cell culture and mouse xenografts, resulting in delayed cell cycle cessation and an elevation in the quantity of memory cells. In contrast to PD-1, which mainly targets secondary co-stimulatory signals through CD28, the mechanism of LAG-3 focuses on limiting TCR signaling. Since PD-1 is more widely expressed than LAG-3, its regulation may involve enhanced regulation of LAG-3 expression through a disintegrin and metalloproteinase (ADAM) mediated proteolytic cleavage. Consequently, LAG-3 may exhibit a lesser efficacy than PD-1 in certain therapeutic contexts. Therefore, to harness the full potential of LAG-3-targeted therapies, further research is imperative to enhance their therapeutic efficacy across a range of diseases, particularly in the realm of autoimmune, inflammatory, and neurological disorders. This approach may enable the development of a next-generation of LAG-3-based therapeutics with improved clinical outcomes 75 .
Other
T-Cell Immunoreceptor With Ig and Immunoreceptor Tyrosine-based Inhibitory Motif Domains
T-cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif (ITIM) domains (TIGIT) is an emerging immune checkpoint molecule that is a corepressor of the immunoglobulin superfamily 76 . TIGIT is expressed on lymphocytes and undergoes upregulation of its expression upon cellular activation. Upon engagement with its specific ligands, TIGIT activates a signaling cascade, which modulates immune cell functionality and the overall immune response, ultimately resulting in an intracellular immunosuppressive response. This mechanism plays a pivotal role in regulating the immune system 77 . Remarkably, besides ligand binding, TIGIT can also perform its immunosuppressive function through the interference of CD226- or CD96-mediated T-cell co-stimulatory signaling78,79. Currently, TIGIT has garnered significant interest as a promising combined target of CAR-T cells for the treatment of hematological malignancies. An array of over 45 TIGIT inhibitors have been developed, predominantly targeted for clinical application in malignant solid neoplasms and leukemia therapies. Nevertheless, only a handful of anti-TIGIT mAbs have advanced to phase III clinical trials to date. Young-Ho Lee et al. 80 pioneered the discovery that the intracellular modulation of PD-1/TIGIT synergistically augments the anti-tumor efficacy of CAR-T cells in a preclinical model. Encouraged by these hopeful preclinical outcomes, a clinical trial evaluating PD-1/TIGIT-knockdown CD19 CAR-T cells is currently underway, aiming to treat patients with R/R DLBCL 80 . In conclusion, the combination of TIGIT and CAR-T cells has promising potential for the treatment of hematological malignancies, and several studies also support the hypothesis that blocking TIGIT can enhance the anti-tumor ability of NK cells, speculating that it could enhance the efficacy of CAR-NK-cell therapy28,81,82. But this strategy still falls short in some respects, such as safety, limited clinical data, and high cost. Subsequent research need to focus on addressing pertinent challenges to advance the further development and clinical application of this therapeutic approach.
Leukocyte Immunoglobulin-Like Receptor Subfamily B
Leukocyte immunoglobulin-like receptor subfamily B (LILRB) is a transmembrane glycoprotein of intracellular immunoreceptor tyrosine inhibitory motifs. LILRB1 and LILRB3 are extensively distributed across a spectrum of hematological malignancies, including AML, leukemia, and B-cell lymphoma. It essentially promotes tumor progression83,84. LILRB4-CAR-T cells were engineered and shown to have potent effects on LILRB4t AML cells both in vitro and in vivo while being nontoxic to normal CD34 T cells 85 . However, LILRB5 in blood system of malignant tumor of the function is not yet clear. Currently, there are no ongoing clinical trials that have yet evaluated the impact of LILRBs in these disorders. Therefore, further scientific research and clinical trials are required for elucidating the specific mechanisms of LILRB5 and its family members in hematologic malignancies.
T-Cell Activation V Domain Ig Inhibitor
VISTA is a structural homolog of PD-L1 that is expressed on both myeloid APCs and Tregs. VISTA promotes the maturation of Tregs and inhibits T-cell activation, VISTA emerges as a structural homologue of PD-L1, exhibiting its presence on both APCs and Tregs. This protein plays a significant role in augmenting the maturation of Tregs while concurrently inhibiting the activation of T cells, thus contributing to immunosuppression of the TME 86 . VISTA and PD-1 inhibit T cells through nonredundant immunomodulatory networks and cooperate to regulate T-cell responses. Inhibiting VISTA increases the recruitment of T cells to the tumor bed, enhances anticancer T-cell function, and further differentiates CD8+ T cells 54 . To date, the sole anti-VISTA mAb (NCT02671955) remains under active clinical investigation. Our comprehension of VISTA is still in its early stages, but this checkpoint displays significant promise in reprogramming various constituents of the TME.
Conclusion
CAR-T-cell therapy and ICIs, as independent strategies for the treatment of hematologic malignancies, have revealed significant therapeutic efficacy. The combination of CAR-T cells and ICIs represents a promising frontier in immunotherapy, addressing two essential elements required for potent immune attack: the presence and functional persistence of immune cells. CAR-T cells promote infiltration, while immune checkpoint blockade ensures sustained persistence and function. In addition, CAR-NK-cell therapy, as an emerging treatment modality, is becoming an important complement to CAR-T-cell therapy. CAR-NK cells possess unique advantages, such as lower immunogenicity and enhanced anti-tumor activity. Recent research advancements have demonstrated that CAR-NK-cell therapy exhibits potent anti-tumor effects both in vitro and in animal models, particularly when combined with immune checkpoint blockade, showing significant therapeutic efficacy and promising potential for clinical application. However, as checkpoint blockade augments the anti-tumor immune reactivity, it also increases the risk of treatment-related side effects, such as CRS and ICANS, necessitating effective management strategies for these severe adverse reactions. Tumor cells may also develop resistance through various mechanisms, including alterations in the expression patterns of immune checkpoint regulators and the secretion of immunosuppressive factors. Despite the limitations associated with CAR-T-cell therapy combined with ICIs, the future utilization of CAR-T-cell therapy in tumor treatment is promising in light of ongoing research and technological advancements. Therefore, future research directions will include the identification of more selective and potent immunosuppressive agents, as well as the formulation of individualized treatment regimens to maximize clinical benefits and minimize the occurrence of adverse reactions.
Footnotes
Acknowledgements
The author(s) gratefully acknowledge the financial supports for this article: This work was partly supported by Medical Science and Technology Project of Zhejiang Province (grant number: 2023KY385) and the Zhejiang Medical Association (grant number: 2015ZYC-A93).
Author Contributions
Manqi Su contributed to the design and conceived of the program and wrote and edited the manuscript. Zhanna Zhang, Panruo Jiang, and Xiaoxia Wang assessed the studies and edited and critically revised the manuscript. Xiangmin Tong and Gongqiang Wu conceived of the program and critically revised the manuscript. All authors read and approved the final manuscript and accept accountabilities for all aspects of this work.
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article, and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by Medical Science and Technology Project of Zhejiang Province (grant number: 2023KY385) and Zhejiang Medical Association (grant number: 2015ZYC-A93).