
Abstract
The HLA-DRB1 gene encodes a protein that is essential for the immune system. This gene is important in organ transplant rejection and acceptance, as well as multiple sclerosis, systemic lupus erythematosus, Addison’s disease, rheumatoid arthritis, caries susceptibility, and Aspirin-exacerbated respiratory disease. The following Homo sapiens variants were investigated: single-nucleotide variants (SNVs), multi-nucleotide variants (MNVs), and small insertions–deletions (Indels) in the HLA-DRB1 gene via coding and untranslated regions. The current study sought to identify functional variants that could affect gene expression and protein product function/structure. ALL target variants available until April 14, 2022, were obtained from the Single Nucleotide Polymorphism database (dbSNP). Out of all the variants in the coding region, 91 nsSNVs were considered highly deleterious by seven prediction tools and instability index; 25 of them are evolutionary conserved and located in domain regions. Furthermore, 31 indels were predicted as harmful, potentially affecting a few amino acids or even the entire protein. Last, within the coding sequence (CDS), 23 stop-gain variants (SNVs/indels) were predicted as high impact. High impact refers to the assumption that the variant will have a significant (disruptive) effect on the protein, likely leading to protein truncation or loss of function. For untranslated regions, functional 55 single-nucleotide polymorphisms (SNPs), and 16 indels located within microRNA binding sites, furthermore, 10 functionally verified SNPs were predicted at transcription factor-binding sites. The findings demonstrate that employing in silico methods in biomedical research is extremely successful and has a major influence on the capacity to identify the source of genetic variation in diverse disorders. In conclusion, these previously functional identified variants could lead to gene alteration, which may directly or indirectly contribute to the occurrence of many diseases. The study’s results could be an important guide in the research of potential diagnostic and therapeutic interventions that require experimental mutational validation and large-scale clinical trials.
Keywords
Introduction
The human leukocyte antigen (HLA) system is the name of the major histocompatibility complex (MHC) in human, generally inherited from parents as a set name haplotype. HLA genes are located on chromosome: 6p (short arm) in the distal portion of the 21.3 band 1 . The HLA system spans a 4 Megabyte (4 × 106 nucleotides) region of the human genome, one of the most polymorphic and gene-dense regions 2 . HLA genes have an important contribution to the immune system and contain several alleles that differ substantially among human populations. The HLA locus has been a focal point of genomic research and clinical practice for several reasons: (1) It is linked to several inflammatory and autoimmune diseases; (2) it is extremely suitable for human genetic diversity studies; and (3) it is critical in tissue and organ transplantation donor–recipient matches 3 . The HLA complex genes and their protein products have been divided into three classes on the basis of their tissue distribution, structure, and function. MHC class II antigens encoded by genes HLA-DM, HLA-DO, HLA-DP, HLA-DQ, HLA-DR loci, and their products are involved in list of the immunoglobulin supergene family4,5. The HLA-DR gene encodes two distinct subunits, DRA (alpha chain) and DRB (beta chain). HLA-DRB1 is a protein-coding gene that belongs to the HLA class II beta chain (approximately 26–28 kDa) paralogs, and it is found on the cell surface2,6.
The HLA-DRB1 gene is located in GRCh38 (Genome Reference Consortium Human Build 38) coordinates 32,578,775 to 32,589,848, has five introns, and is encoded by six exons. Exon 1 encodes the leader peptide; exons 2 and 3 encode the two extracellular domains; exon 4 encodes the transmembrane domain; and exon 5 encodes the cytoplasmic tail 7 (https://www.ncbi.nlm.nih.gov/gene/3123). Compared with its paralogs DRB3, DRB4, and DRB5, DRB1 is expressed at a level that is five times higher 8 . HLA genes region is the most polymorphic in the human genome, and the HLA-DRB1 gene is the most polymorphic in class II of this system9,10. The HLA-DRB1 locus had 3,196 alleles in May 2022, according to the IPD-IMGT/HLA database 11 (https://www.ebi.ac.uk/ipd/imgt/hla/about/statistics/). Many HLA-DRB1 alleles (a gene’s variant forms) have been associated with various diseases. HLA- DRB1*1501 12 , 13 , DRB1*03 14 , DRB1*0404 15 , DRB1*04:05 16 , DRB1*13 17 , and DRB1*04 18 , 19 alleles have been associated with multiple sclerosis12,13, systemic lupus erythematosus 14 , Addison’s disease 15 , rheumatoid arthritis 16 , caries susceptibility 17 , graft survival in organ transplant recipients 18 , and Aspirin-exacerbated respiratory disease 19 . The 1,000 genome project revealed that single-nucleotide polymorphisms (SNPs) account for the majority of human genetic variation 20 .
SNPs are single-nucleotide variants (SNVs) in DNA sequence with a population allele frequency of 1% or higher. It normally occurs throughout the genome with the frequency of about one of each 600 to 1,000 nucleotide, which is considered the simplest and common type of genetic marker leading to DNA variation among individuals21,22. Non-synonymous SNPs (nsSNP) are a type of SNP that represents amino acid substitutions and protein variations in humans. Previous research indicates that nsSNPs account for roughly half of the mutations involved in various genetic diseases 23 . Other important types of genomic variation are indels, which are insertions or deletions of one or more nucleotides in the DNA sequence 24 .
The SNP Database (dbSNP) is one of the NCBI’s subdivided databases that contain human single-nucleotide variations, microsatellites, and small-scale insertions and deletions. SNP database contains 1,076,992,604 Homo sapiens variants as of May 28, 2022. There were 957,193,110 SNPs, SNVs, or MNVs (multi-nucleotide variants) among the total number of variants, and 29,620,962 Indels (single or small length insertions–deletions). (https://www.ncbi.nlm.nih.gov/snp/). Functional variants within coding regions may affect protein structure and function, whereas non-coding variants may have an impact on protein expression25,26. Pathological non-coding variants could have an alteration role in various regulatory functions within the genome, such as interacting with transcription factors (TFs), and microRNA (miRNA) 27 . Identification of variants responsible for phenotypic changes is considered difficult, as it necessitates multiple tests for different variants in candidate genes8,27,28. One possible solution would be to prioritize variants based on their structural and functional significance using various bioinformatics prediction tools. The use of computational methods for gaining biological insight is well established29–33. Thus, the current study aimed to in silico analyze all human SNVs, MNVs, and short Indels in the HLA-DRB1 gene’s coding and untranslated regions to significantly predict functional variants that could affect gene expression and protein product function/structure.
Materials and Methods
Variants Dataset
HLA-DRB1 gene variants were discovered using the NCBI SNP database (https://www.ncbi.nlm.nih.gov/SNP/) on April 14, 2022. The HLA-DRB1 variants (SNPs, SNVs, MNVs, and INDELs) were retrieved from the SNP database build 155 and mapped on genome assembly GRCh38 using Variation Viewer (https://www.ncbi.nlm.nih.gov/variation/view/). Variants in coding and 3′/5′ untranslated regions have been identified for computational analysis of their effect(s). Several tools have been used to improve the accuracy and reliability of identifying pathogenic variants and their effects on the structure, function, and expression of HLA-DRB1 (Fig. 1).

Flowchart for the in silico analysis of variants in the HLA-DRB1 gene and their biological consequences. The black shapes represent the type of data, while the blue shapes represent the names of the prediction tools. SNP: single-nucleotide polymorphism; SNV: single-nucleotide variant; MNV: multi-nucleotide variant; INDEL: insertion–deletion; SIFT: Sorting Intolerant From Tolerant; PANTHER: Protein Analysis Through Evolutionary Relationships; GO: Gene Ontology; PROVEAN: Protein Variation Effect Analyzer.
Coding Variants Analysis (nsSNPs/nsSNVs, Indels, Stop Gain, and MNVs)
To identify the most deleterious missense or nsSNVs, seven distinct bioinformatics tools, namely, SIFT (Sorting Intolerant From Tolerant), PolyPhen, PredictSNP, Panther (Protein Analysis Through Evolutionary Relationships), SNP&GO (Gene Ontology), PROVEAN (Protein Variation Effect Analyzer), and SNAP2, have been used34–40. All nsSNVs identified as harmful by the previous seven tools and predicted as instabilities by the I-mutant server are categorized as high risk (Table 2) 41 . Among the total high-risk variants, nsSNVs with high evolutionary conservation and located in domain sites were chosen (Table 4). InterPro database and the Consurf server were used to identify domains and high evolutionary conservation (grade ≥ 6) amino acids (Table 3 and Fig. 2)42,43. To understand the effect of nsSNVs on protein structure, HOPE tool using sequence and missense-3D server using structure model were used (Tables 5 and 6 and Fig. 3)44,45. The related protein sequence was (accession number: P01911) obtained from Uniprot database (http://www.uniprot.org). Phyre2 and Swiss-Model servers were used to predict the protein models46,47. To select the high-quality model, two evaluation tools [PSICA (Protein Structural Information Conformity Analysis) and ModFOLD8] were used (Figs. 4 and 5)48,49. For more investigation in the coding regions, indel was entered into the SIFT algorithm to anticipate their functional effect (Table 7). Furthermore, SNVs/indels that result in a premature stop codon (stop gain) and MNVs were submitted to Variant Effect Predictor to assess the impact of this change (Table 8) 50 ; https://www.ensembl.org/info/docs/tools/vep/index.html. The ProtParam server was then used to assess the impact of conserved and domain-located nsSNVs on protein physicochemical parameters (Table 9) 51 .

Evolutionary conservancy of HLA-DRB1 produced by Consurf server.

Structural alteration by HOPE server. The protein is shown in gray, the wild type residue in green, and the mutant residue in red.

Protein models evaluation using PSICA server. The illustration on the left represents the PHYRE2-server model, while the right represents the SWISS-MODEL structure. PSICA: Protein Structural Information Conformity Analysis.

Protein models evaluation using ModFOLD8. The illustration on the left represents the PHYRE2-server model, while the right represents the SWISS-MODEL structure. The upper number represents the global model quality score, while the lower represents the confidence and P value.
SIFT server
Make an alignment between an order sequence with a large number of homologous sequences to predict if an amino acid substitution will have a phenotypic effect. The Residual’s score ranges from zero to one. If the score is less than or equal to 0.05, the amino acid substitution is predicted to be harmful; if the score is greater than 0.05, the substitution is tolerated 34 ; https://sift.bii.a-star.edu.sg/.
PolyPhen-2 (Polymorphism Phenotyping v2) server
A tool uses simple physical and comparative considerations to predict the impact of an amino acid substitution on the structure and function of a human protein. A mutation is classified qualitatively, as benign, possibly damaging, or probably damaging 35 . http://genetics.bwh.harvard.edu/pph2/.
PredictSNP tool
The server was developed by combining six disease-related mutation prediction programs. The predicted effect is color-coded: Neutral mutations are green, while deleterious mutations are red 36 ; https://loschmidt.chemi.muni.cz/predictsnp/.
PANTHER (Protein Analysis Through Evolutionary Relationships)
This classification system was designed to classify proteins (and their genes) to facilitate high-throughput analysis. Proteins have been classified according to family/subfamily, molecular function, and biological process. The tool assesses the functional effects of nsSNPs, with three possible outcomes: probably benign, possibly damaging, and probably damaging 37 . PANTHER computes the length of time (in millions of years) that a given amino acid has been preserved in the lineage that led to the protein of interest. The longer the preservation time, the more likely it is that functional impact will occur. The method is known as PANTHER-PSEP (position-specific evolutionary preservation). The preservation time outputs are classified as >450, between 200 and 450, and <200 million years, corresponding to probably damaging, possibly damaging, and probably benign 37 ; http://www.pantherdb.org/tools/csnpScoreForm.jsp
SNP&GO (Gene Ontology)
The server is based on Support Vector Machines (SVM) and has been optimized to predict if a given single-point protein variation can be classified as disease-associated or neutral 38 ; https://snps.biofold.org/snps-and-go/snps-and-go.html
PROVEAN (Protein Variation Effect Analyzer)
It is a software tool that predicts whether an amino acid substitution or indel will affect a protein’s biological function. PROVEAN can be used to filter sequence variants to identify non-synonymous or indel variants that are predicted to be functionally important 39 . The PROVEAN prediction score classifies the substitution as having a deleterious or neutral effect on protein function; http://provean.jcvi.org/index.php
SNAP2 server
A trained classification algorithm based on a machine learning device known as a neural network. SNAP2 predicts the impact (effect) of single amino acid substitutions on protein function. The prediction score ranges from –100 (strong neutral) to +100 (strong effect). According to the findings, the prediction score is to some extent correlated to the severity of effect 40 ; https://www.rostlab.org/services/snap/
I-mutant server
I-Mutant v3.0 is a suite of SVM-based predictors integrated in a unique web server. It offers the opportunity to predict the protein stability changes upon single-site variations from the protein structure or sequence. The I-mutant result is either decrease/increase stability or neutral 41 ; http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi
InterPro database
It performs functional protein analysis by categorizing them into families and predicting domains and key locations. InterPro employs prediction models, known as signatures, offered by several databases to categorize proteins in this manner 42 ; https://www.ebi.ac.uk/interpro/
Consurf server
It is a bioinformatics tool that uses phylogenetic relationships between homologous sequences to estimate the evolutionary conservation of amino/nucleic acid positions in a protein/DNA/RNA molecule. Position-specific conservation scores are computed using the empirical Bayesian or ML algorithms. For illustration, the continuous conservation scores are grouped into nine categories, ranging from the most changeable places (grade 1) in turquoise to the most conserved positions (grade 9) in maroon 43 ; https://consurf.tau.ac.il/
HOPE server
An automatic mutant analysis server can provide information about a mutation’s structural effects. HOPE gathers information from a wide variety of sources. Data are stored in a database and used in a decision scheme to determine the effects of a mutation on the protein’s 3D structure and function. HOPE’s final report includes discovered data on contacts (metal, DNA, hydrogen bonds, ionic interactions, etc.), structural locations (motifs, domains, transmembrane domains, etc.), non-structural features (post-translational modifications), known variants at that position, and amino acid physicochemical properties (size, charge, and hydrophobicity). HOPE creates an easy-to-use and understandable report with text, figures, and animations 44 ; https://www3.cmbi.umcn.nl/hope/
Phyre2 and Swiss-Model tools
Protein structure prediction automated servers. Both algorithms are based on comparative modeling methods. Phyre2 uses the alignment of hidden Markov models via HHsearch to significantly improve accuracy of alignment and detection rate. Phyre2 also could use AB-initio method to determine the tertiary structure of protein in the absence of experimentally solved structure46,47; http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index, https://swissmodel.expasy.org/.
PSICA and ModFOLD v.8
Both are protein structure quality assessment servers. PSICA is the official implementation of MUfoldQA_S and MUfoldQA_C methods. It is designed to evaluate how much a tertiary model of a given protein primary sequence conforms to the known protein structures of a similar protein 48 . ModFOLD8 combines the strengths of multiple pure-single and quasi-single model methods to predict global and local quality of 3D protein models. The global model quality scores range between 0 and 1. In general, scores less than 0.2 indicate there may be incorrectly modeled domains and scores greater than 0.4 generally indicate more complete and confident models, which are highly similar to the native structure. Depending on the P value, each model is also assigned a score confidence level. CERT, HIGH, MEDIUM, LOW, and POOR are the confidence levels from best to worst 49 ; http://qas.wangwb.com/~wwr34/mufoldqa/index.html, https://www.reading.ac.uk/bioinf/ModFOLD/.
Missense3D tool
It predicts the structurally damaging change in the mutant structure 45 ; http://missense3d.bc.ic.ac.uk/~missense3d/
ProtParam server
A program that calculates various physical and chemical parameters for a protein sequence. Manual variants were applied to the reference protein sequence separately and resubmitted to calculate the properties changed by variant to detect the impact of the nsSNVs. The calculated parameters include the molecular weight, theoretical pI, atomic composition, extinction coefficient, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) 51 .
Untranslated Regions Variants Analysis (SNPs/SNVs and INDELs)
The PolymiRTS database and the SNP Function Prediction tool were used to predict functional variants based on genetic changes (SNPs/SNVs and INDELS) within 3′/5′ UTRs of the HLA-DRB1 gene (Table 10). PolymiRTS (Polymorphism in microRNAs and their Target-Sites) is a database of naturally occurring DNA variations in the seed regions and target sites of miRNAs. SNPs and INDELs in miRNAs and their target sites may have an impact on miRNA-mRNA interaction, and thus miRNA-mediated gene repression 52 . SNP Function Prediction (FuncPred) was used to predict the effect of SNVs/indels at transcription factor-binding sites (TFBSs; Table 11). Functional variants in the previous region may affect gene expression level, location, or timing 53 ; https://compbio.uthsc.edu/miRSNP/, https://snpinfo.niehs.nih.gov/snpinfo/snpfunc.html.
Gene–Gene and Protein–Protein Interactions
GeneMANIA server employed a vast number of functional association data to build a biological network interaction of the top 20 genes associated with our HLA-DRB1 target gene. GeneMANIA uses a guilt-by-association approach to identify the most related genes to a query gene set. Protein and genetic interactions, pathways, co-expression, co-localization, and protein domain similarity are all examples of association data 54 . Inbio Discover was utilized to establish high confidence protein–protein interactions (PPIs) network. The inBio-Map, a comprehensive map of human protein biology with over 6 million traceable entries, is used by InBio Discover. The predicted trusted interaction networks are based on experimental evidence, pathways, and other curated resources 55 ; https://genemania.org/, https://inbio-discover.com/.
Results
Within the data retrieval date, the HLA-DRB1 gene contained a total of 9,648 variants, including 7,159 SNVs and 1,078 indels. Except for one, none of the variants have been registered to be significantly associated with human disease, according to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). In addition, only 26 variants have related publications. From the total variation data, various variants within coding and untranslated regions were chosen for the current study. Information on selected variants is shown in Table 1.
Distributions of SNVs/MNVs and INDELs.

SNV: single-nucleotide variant; MNV: multi-nucleotide variant; INDEL: insertion–deletion.
Seven different tools (SIFT, PolyPhen, PredictSNP, PANTHER, SNP&GO, PROVEAN, and SNAP2) with different prediction algorithms were used to identify nsSNVs with significant deleterious effects that could affect the biological structure and function of HLA-DRB1 protein. Out of 375, 91 nsSNVs were predicted by all previous tools to be functional (deleterious or damaging). The I-mutant server predicted changes in stability for all 91 functional nsSNVs identified. Following all previous analyses, the 91 nsSNVs were classified as “high-risk” (Table 2). Most of the high-risk variants are located in exon 2.
High-Risk nsSNPs Identified by Seven In Silico Programs and Their Impact on Protein Stability Effect.
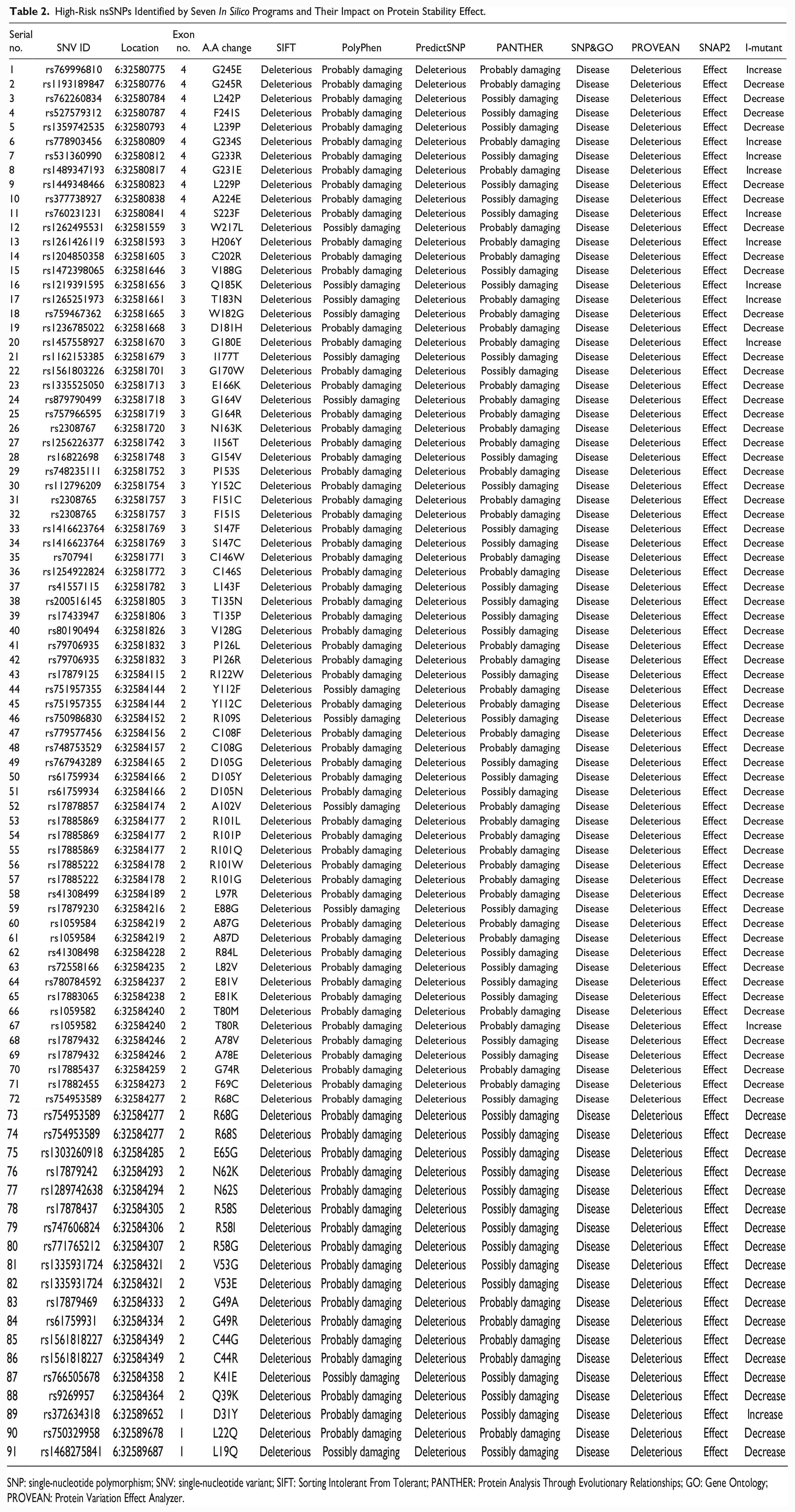
SNP: single-nucleotide polymorphism; SNV: single-nucleotide variant; SIFT: Sorting Intolerant From Tolerant; PANTHER: Protein Analysis Through Evolutionary Relationships; GO: Gene Ontology; PROVEAN: Protein Variation Effect Analyzer.
The Consurf server and the InterPro database were used to predict the effects of evolutionarily conserved variants on protein functions. The conservation analysis of the HLA-DRB1 protein predicted that 154 positions (≥6 scores) out of 266 amino acids were conserved, as seen in Fig. 2. Table 3 includes the locations and domain names of the InterPro resource that were found. Among the high-risk variants, 25 nsSNVs were identified as conserved and located in domain regions, and they may disrupt or abolish domain function (Table 4).
Predicting Domains Using InterPro.

Non-Synonymous SNPs That Are Highly Conserved and Located in Domains’ Sites.

The symbol “—” refers to unavailable data.
CLIN/SIG: clinical significance refers to the ClinVar database, which compiles data on genomic variation and its impact on human health; SNP: single-nucleotide polymorphism.
The effects of the 25 nsSNVs on protein structure were predicted using two tools. The first is the HOPE server, which predicts structural effects based on protein sequence, and the second is Missense3D, which uses a protein model to predict effects. HOPE outcomes show the change in amino acid physiochemical properties, effects on their location, and may disturb the core structure of the located domain (Table 5 and Fig. 3). SWISS-MODEL and PHYRE tools predicted two HLA-DRB1 protein models. Following evaluation by PSICA and ModFOLD, the PHYRE model was selected (Figs. 4 and 5). Using the Missense3D tool, 13 nsSNVs were predicted to cause structural damage to the protein model. The discovered structural damage is displayed in Table 6.
HOPE-Based Protein Sequence Predictions (Structural and Function Change).

The symbol “—” refers to unavailable data.
W: wild type residue; M: mutant type residue.
Structural Modifications Brought About by an Amino Acid Substitution Using Missense3D Tool.

Other types of variants (MNVs and indels) were analyzed for further analysis within coding regions to determine whether they might have a harmful effect on protein. All MNVs showed no significance damage appears. In contrast, 31 out of 36 indels were predicted as harmful by SIFT (Table 7). In addition, within the coding sequence (CDS), 23 stop-gain variants (SNVs/INDELs) were predicted as high impact (Table 8). Last, all nsSNVs demonstrated changes in overall protein physicochemical parameters. The properties changed by all 25 conserved and domain-located high-impact nsSNVs were molecular weight, atomic composition, and GRAVY (Table 9).
SIFT Server Functional Prediction of All Indels in Coding Regions.

SIFT: Sorting Intolerant From Tolerant.
High-Impact Stop-Gain SNVs/INDELs Identified by Variant Effect Predictor.
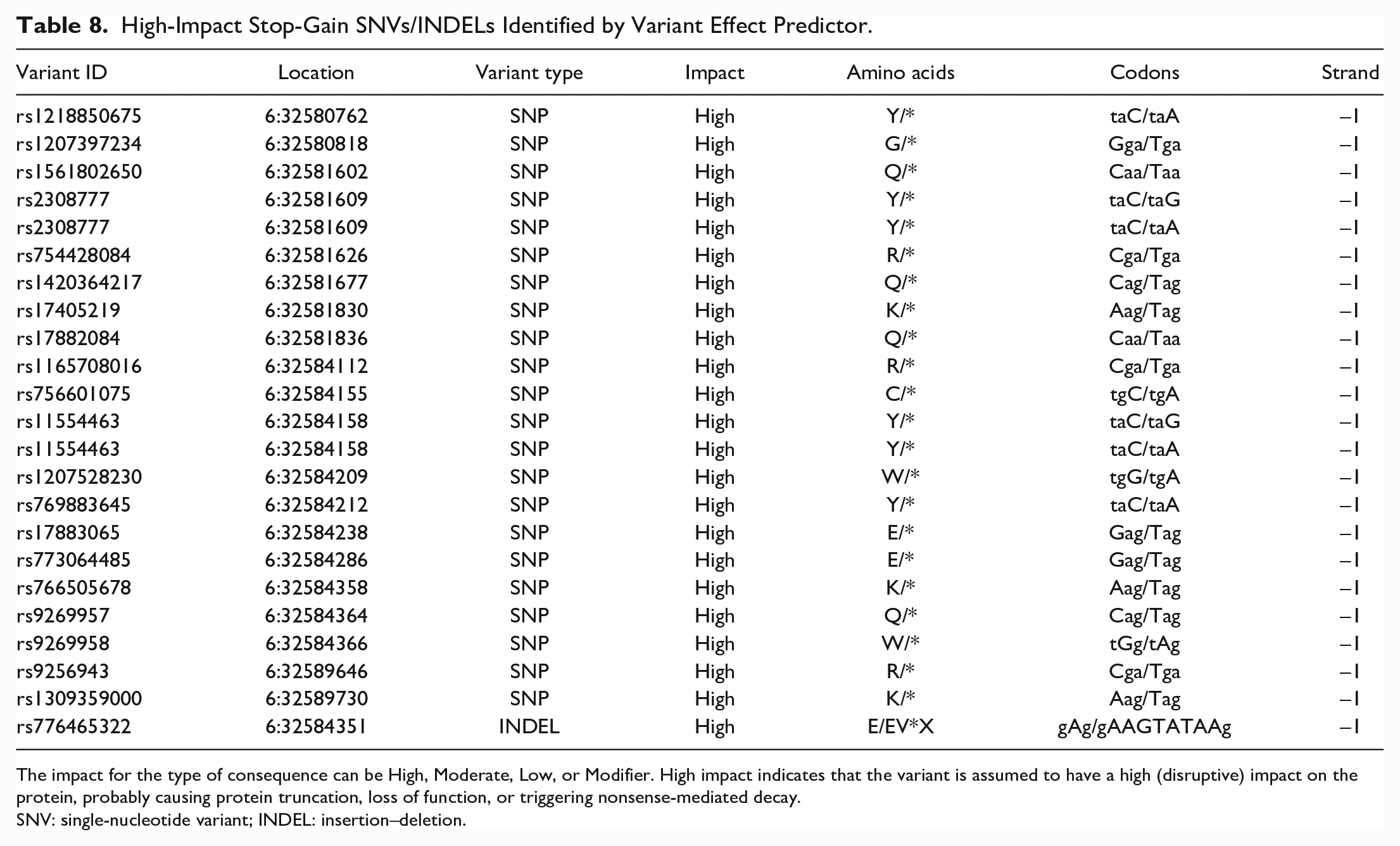
The impact for the type of consequence can be High, Moderate, Low, or Modifier. High impact indicates that the variant is assumed to have a high (disruptive) impact on the protein, probably causing protein truncation, loss of function, or triggering nonsense-mediated decay.
SNV: single-nucleotide variant; INDEL: insertion–deletion.
The Effect of nsSNVs on HLA-DRB1′ Protein Physicochemical Parameters.

The accession number for the reference sequence is P01911 (https://www.uniprot.org/). Total –ve: total negatively charged residues. Total +ve: total positively charged residues. The parameters that have been changed compared with the reference are highlighted in bold.
SNV: single-nucleotide variant; HLA: human leukocyte antigen; GRAVY: grand average of hydropathicity index.
The purpose of analyzing variants in untranslated regions is to predict the effects of variants in miRNAs and TFBSs. Functional variants (SNVs and indels) within previous regions could affect gene expression. The results of PolymiRTS Database show that 16 indels and 55 SNPs in the 3′UTR have functional effects on various miRNA binding sites (Table 10). Furthermore, no indels and 10 functionally verified SNPs (of 5′UTR variants) were predicted to affect the activity of TFBSs. The findings are summarized in Table 11. GeneMANIA was used to construct the gene–gene interaction network of the HLA-DRB1 target gene and the closest 20 genes (Fig. 6). Thus, to gain a better understanding, a network of PPIs was constructed using the inBio-Map resource (Fig. 7). The PPIs network that was built predicted 25 interacted proteins and 44 interactions.
Functional SNPs/Indels in the 3′UTR.
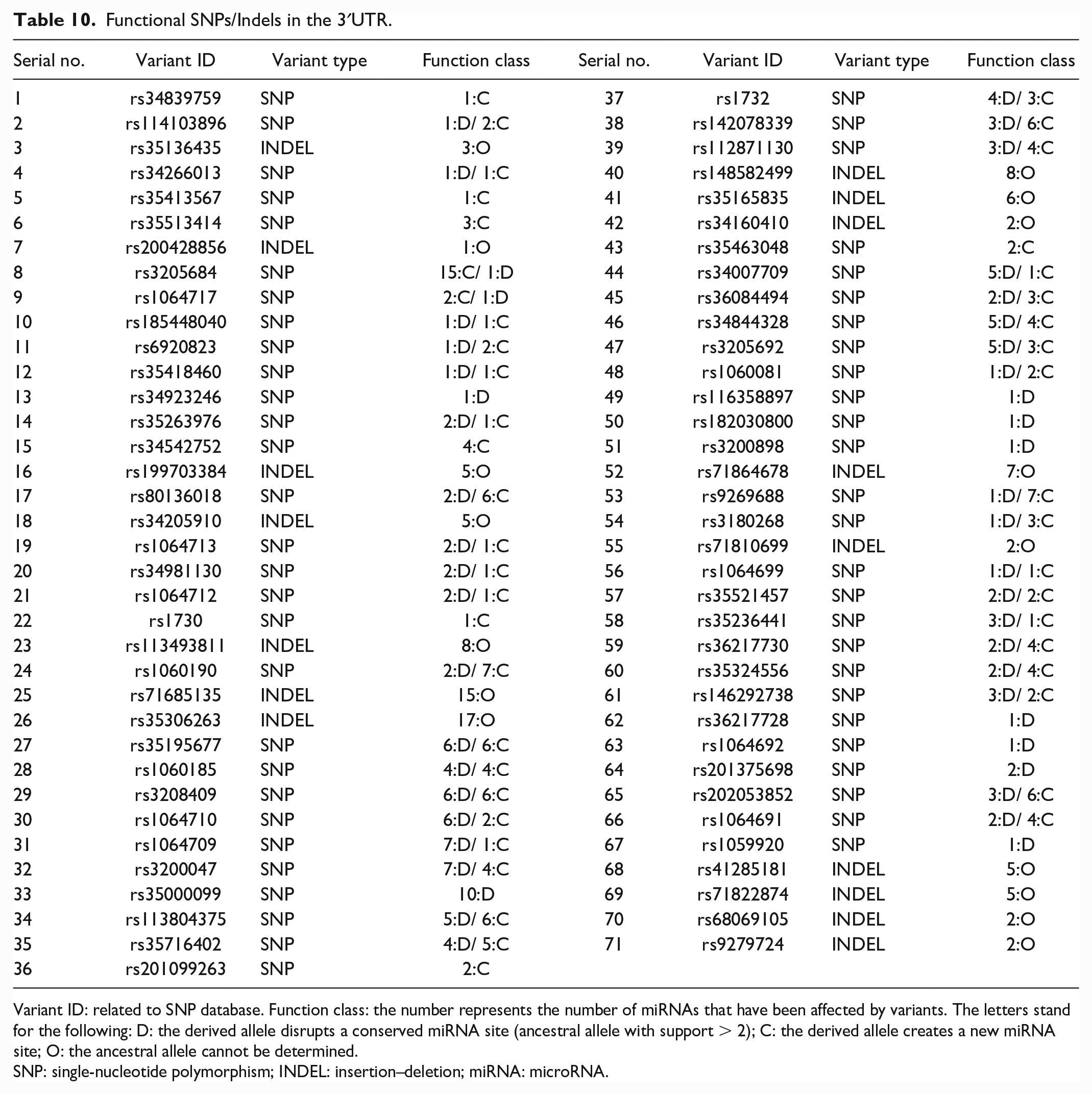
Variant ID: related to SNP database. Function class: the number represents the number of miRNAs that have been affected by variants. The letters stand for the following: D: the derived allele disrupts a conserved miRNA site (ancestral allele with support > 2); C: the derived allele creates a new miRNA site; O: the ancestral allele cannot be determined.
SNP: single-nucleotide polymorphism; INDEL: insertion–deletion; miRNA: microRNA.
Functionally Verified SNPs at a Transcription Factor-Binding Site.

For more information on regulatory potential and conservation scores, see https://snpinfo.niehs.nih.gov/snpinfo/guide.html#snpfunc.
SNP: single-nucleotide polymorphism.

Gene–gene interaction network of the HLA-DRB1 gene predicted by GeneMANIA.

Protein–protein interaction network of the HLA-DRB1 protein predicted by inBio-Discover. Pathway interactions are shown as blue lines, remaining interactions are inBio-Map high-confidence interactions.
Discussion
HLA-DRB1 gene and its product protein are important in several inflammatory diseases, autoimmune diseases, genetic diversity, and tissue or organ transplantation donor–recipient matches 3 . The protein generated by the HLA-DRB1 gene, known as the beta chain, connects (binds) to another protein produced by the HLA-DRA gene, known as the alpha chain. They combine to produce the HLA-DR antigen-binding heterodimer, a functional protein complex. This complex presents foreign peptides to the immune system to activate the body’s immunological response 6 . Variations in the structural conformation of the HLA-DRB1 protein during bio-molecular interactions are critical for its function. Therefore, determining the effects of harmful HLA-DRB1 variants and their association with various diseases is critical. The purpose of this study was to use computational analysis to identify the most harmful variants (SNVs, MNVs, and INDELS) and their effects on the HLA-DRB1 structure, function, and expression.
In terms of substitution single-variants, several tools predicted that 91 missense (nsSNV) and 22 stop-gain variants within coding regions were functional. The 22 stop-gain variants were classified as high impact, implying that the variant will have a significant (disruptive) effect on the protein, most likely resulting in protein truncation or loss of function. The 91 nsSNVs were classified as high risk after the target protein’s stability changed. Thirteen of the high-risk nsSNVs (rs9269957, rs17879469, rs17879242, rs17879432, rs17885437, rs17883065, rs41308498, rs1059584, rs17879230, rs41308499, rs17885869, rs61759934, and rs41557115) correspond to pathological variants predicted by Hassan et al. 56 The variants identified as pathological by Hassan’s discovery but not in this study could be due to the increased number of tools used in the current study. The update to the SNP and tool databases may have caused the vice versa to occur. The Consurf server and the InterPro database were used to predict the effects of evolutionarily conserved variants that are located in domains. InterPro resource integrates signatures from the following 13 member databases: CATH, CDD, HAMAP, MobiDB Lite, Panther, Pfam, PIRSF, PRINTS, Prosite, SFLD, SMART, SUPERFAMILY, and TIGRfams. Among the high-risk variants, 25 nsSNVs were identified as conserved and located in domain regions, and they may disrupt or abolish domain function. The effects of the 25 nsSNVs on protein structure were predicted based on sequence and model using HOPE and Missense3D, respectively. The HOPE results revealed that the amino acid properties of the 25 nsSNVs changed and have the potential to disrupt the domain’s core structure. Two algorithms (SWISS-MODEL and PHYRE) were used to predict the HLA-DRB1 models to use the Missense3D tool. Following evaluation by PSICA and ModFOLD, the PHYRE model was selected. Several factors contributed to the selection of the PHYRE model, including its coverage of the entire protein (266 amino acids), higher overall quality scores, and best confidence value. The Missense3D tool predicted that 13 of the 25 nsSNVs would cause structural damage to the protein model.
Additional types of variants (MNVs and indels) were analyzed for further analysis within coding regions to determine whether they might have a harmful effect on protein function. All MNVs showed nil significance damage appears. In contrast, SIFT predicted 31 indels to be harmful, while the Variant Effect Predictor predicted only one to lead to premature protein. Functional predicted Indels might affect a few numbers of amino acids and even the complete protein as shown. According to physiochemical properties, the HOPE tool, as previously mentioned, revealed differences in the level of residues between wild and new types, whereas ProtParam indicated that variants caused changes in the entire protein. All 25 conserved and domain-located high-impact nsSNVs agreed to alter the protein’s molecular weight, atomic composition, and GRAVY, but there is a divergence in other properties. In general, high-risk nsSNVs affect protein structure, function, and physicochemical properties.
The goal of analyzing variants (SNVs and indels) in 3′/5′ untranslated regions is to predict the effects of variants that may affect the level, location, or timing of gene expression using PolymiRTS and SNP Function Prediction tools 53 . The 3′UTR of the messenger RNAs that serve as their targets is where miRNAs bind 57 . The PolymiRTS Database revealed that the 16 indels and 55 SNPs have functional effects on various miRNA binding sites. Previous variants disrupted conserved sites of 131 miRNAs and created new binding sites for 149 miRNAs. Furthermore, no indels and 10 functionally verified SNPs (of 5′UTR variants) were predicted to affect transcriptional regulation by influencing the activity of TFBSs.
Genetic interaction is the set of functional association between genes. Gene interactions occur when two or more allelic or non-allelic genes of same genotype influence the outcome of particular phenotypic characters. To understand the molecular basis of this complex biological phenomenon, there is a need of genetic interaction mapping where the effects on one gene are modified by one or several other genes. The gene–gene interaction network of the HLA-DRB1 target gene and the closest 20 genes was built using GeneMANIA. A potent tool for systematically defining gene function and pathways is mapping genetic interactions, accomplished by simultaneously perturbing pairs of genes that report how genes interact with one another 58 . A case of extreme genetic interaction is synthetic lethality, in which two mutations combine to create a lethal double mutant phenotype even though neither of them would be fatal on their own 59 . Most proteins work consecutively with other proteins in living organisms. Thus, PPI studies give crucial information for comprehending the complicated biological processes that occur in live cells 60 . Thus, to gain a better understanding, a network of PPIs was constructed using the inBio-Map resource. Deleterious variants in the HLA-DRB1 protein could disrupt its interaction with confidence interaction proteins.
Conclusion
HLA-DRB1 gene plays an important role in organ transplantation rejection and many other diseases. The current study shows the in silico analysis of genetic variants within the coding region, and 3′/5′ UTRs. Pathological variants may have a direct or indirect impact on the intramolecular/intermolecular interactions of amino acid residues, protein expression, and disease risks. We discovered significant structural and functional changes in HLA-DRB1 proteins by analyzing the conformational changes and interactions of amino acid residues. These changes can explain the activity deviations caused by several variants. This is the first study to predict the effects of coding and 3′/5′ UTR variants (SNVs, MNVs, and indels) in the HLA-DRB1 gene. The findings demonstrate that employing in silico methods in biomedical research is extremely successful and has a major influence on the capacity to identify the source of genetic variation in diverse disorders. The study’s results could be an important guide in the research of potential diagnostic and therapeutic interventions that require experimental mutational validation and large-scale clinical trials.
Footnotes
Ethical Approval
Ethical approval is not applicable for this article.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.