
Abstract
Head and neck squamous cell carcinoma (HNSCC) is a malignant tumor of the upper aerodigestive tract affecting the oral cavity, lips, paranasal sinuses, larynx, and nasopharynx. Proteogenomics combines proteomics and genomics and employs mass spectrometry and high-throughput sequencing technologies to identify novel peptides. The aim of this study was to identify potential protein biomarkers for clinical treatment of HNSCC. To achieve this, we utilized two sets of data, one on proteins from The Cancer Proteome Atlas (TCPA) and the other on gene expression from The Cancer Genome Atlas (TCGA) database, to evaluate dysfunctional proteogenomics microenvironment. Univariate Cox regression analysis was performed to examine the relationship between protein signatures and prognosis. A total of 19 proteins were significantly associated with overall survival (OS) of patients, of which E2F transcription factor 1 (E2F1; HR = 4.557, 95% CI = 1.810 to 11.469) and enhancer of zeste homolog 2 (EZH2; HR = 0.430, 95% CI = 0.187 to 0.984) were the most differentially expressed between patients with longer and shorter OS, respectively. Furthermore, multivariate Cox regression analysis on six proteins (ERALPHA, HER3, BRAF, P27, RAPTOR, and E2F1) was performed to build the prognostic model. The receiver operating characteristic curves were used to determine whether the expression pattern of survival-related proteins could provide an early prediction of the occurrence of HNSCC. Herein, we found an AUC of 0.720. Based on an online database, we identified novel protein markers for the prognosis of HNSCC. The findings of the present study may provide new insights into the development of new and reliable tools for precise cancer intervention.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is a malignant tumor of the upper aerodigestive tract affecting the oral cavity, lips, paranasal sinuses, larynx, and nasopharynx 1 . Currently, it is the sixth most prevalent cancer globally and accounts for more than 650,000 new cases and 90,000 deaths annually 2 . Known risk factors for the disease include smoking cigarettes, infection with human papillomavirus (HPV), and alcohol consumption 3 . One of the main challenges in HNSCC treatment is late diagnosis because symptoms tend to occur in the advanced stages of the disease. Consequently, the 5-yr survival rate of individuals with HNSCC is less than 50% and can drop to less than 35% in patients that develop local recurrence or metastasis 4 . Once in the advanced stages, treatment interventions can affect the structure and function of organs involved in speech, taste, smell, swallowing, as well as other vital organs of the body. These eventualities reduce the overall quality of life of the affected individuals 5,6 .
Proteogenomics is a field of research that combines proteomics and genomics and utilizes mass spectrometry and high-throughput sequencing technologies to identify novel peptides. Mass spectrometry–based matching or identification of homologous peptides provides evidence of gene expression at protein level 7 . As such, it allows in-depth analysis of interactions between host and respective pathogens. Combining advanced genomics with proteomics has the potential of revealing useful biomarkers that can drive the development of new diagnostic techniques and therapies. Many studies have explored the role of proteogenomics in assessing the prognosis of cancers, but even so, the mechanism with which tumors progress remains unclear. Ma et al. 8 found that C-C motif chemokine ligand protein mediates progression and angiogenesis of Epstein Barr virus (EBV)–mediated nasopharyngeal carcinoma. Moreover, patients with oral squamous cell carcinoma exhibit overexpressed CD44 molecule S100A7, and S100P proteins. Consistently, these proteins are considered as potential biomarkers for early detection of malignant tumors 9 . A few integrative tools for complete proteogenomics analyses have been developed; however, there are currently no high-throughput technologies for evaluating protein biomarkers that can predict the overall survival (OS) or response to HNSCC immunotherapy. Because high-throughput expression data are available, it is feasible to use global information on gene expression to analyze the association between genomics and prognoses of tumors. In this study, we utilized two sets of data, one on proteins, from The Cancer Proteome Atlas (TCPA), and the other on gene expression, from The Cancer Genome Atlas (TCGA) database, to evaluate dysfunctional proteogenomics microenvironment with an aim of identifying useful protein biomarkers for clinical treatment of HNSCC.
Materials and Methods
Data Collection
Data was collected from TCPA, which contained a protein profile of 8,167 tumor samples. These proteins were primarily reflective of the gene expression of genetic maps in TCGA database. They provide a unique opportunity to validate the reliability of data in TCGA and identify model cell lines for functional research 10 . TCGA has generated many platforms for data on cancer genomics as well as proteomics using the Reverse Phase Protein Array, which measures the level of about 150 proteins and 50 phosphoproteins in tumors 11 . Data on proteomics was therefore downloaded from TCPA (level 4), together with gene expression profiles from TCGA.
Establishing Prognostic Related Gene Signatures
Univariate Cox regression analysis was used to identify the prognostic genes and establish genetic characteristics. The gene signature was calculated as risk score = (CoefficientmRNA1 × expression of mRNA1) + (CoefficientmRNA2 × expression of mRNA2) + . + (CoefficientmRNAn × expression mRNAn). Based on the median, each parameter was categorized into a low-risk and a high-risk group. Kaplan–Meier survival analysis was performed to compare the survival rate between the study group and the control group.
Establishing a Predictive Nomogram
Nomograms are often used to determine the prognosis of cancer because they can simplify statistical prediction models and provide individualized numerical assessment of the likelihood of an event (such as relapse or death) 12 . In the present study, a receiver operating characteristic (ROC) curve was plotted to evaluate the accuracy of the proteins as prognostic markers for HNSCC. Univariate and multifactorial Cox regression analyses were performed to compare the association between the expression of specific proteins and clinicopathological variables. Data with missing values were deleted from the survey. These data were analyzed using R packages in R (version 3.5.3 http://www.r-project.org/). Statistical significance regarding Perl language for data matrix and all data processing was set at P < 0.05.
Results
Prognostic Protein Signatures
Data on proteomics were first retrieved from TCPA. The corresponding genomic data on clinical survival was downloaded from TCGA database. Univariate Cox regression analysis identified 19 proteins that were significantly associated with OS of patients, of which E2F transcription factor 1 (E2F1; hazard ratio (HR) = 4.557, 95% confidence interval (CI) = 1.810 to 11.469) and enhancer of zeste homolog 2 (EZH2; HR = 0 .430, 95% CI = 0.187 to 0.984) were the most differentially expressed between patients with longer and shorter OS, respectively (Table S1). The volcano plot was drawn to visualize the expression profile of survival-related proteins. The red and green dots indicate high-risk and low-risk proteins, respectively (Fig. 1A). Multivariate Cox regression analysis on the six proteins Estrogen receptor alpha (ERALPHA), Erb-b2 receptor tyrosine kinase 3 (HER3), B-Raf proto-oncogene serine/threoninekinase (BRAF), Cyclin-dependent kinase inhibitor p27 (P27), mTOR regulation-related protein (RAPTOR), and E2F transcription factor 1 (E2F1) was performed to build the prognostic model. The formula for our model was: Risk Score = 0.84 × ERALPHA + 0.89 × HER3 + (–0.64) × BRAF + (–0.86) × P27 + 1.79 × RAPTOR + 1.33 × E2F1. Significant differences in expression levels of ERALPHA (P = 2.934e−12), HER3 (P = 3.779e−07), BRAF (P = 7.187e−08), P27 (P = 1.059e−06), RAPTOR (P = 2.694e−11), and E2F1 (P = 6.311e−12) were found between the high-risk and the low-risk groups.

(A) Volcano plot showing the survival-related proteins (red dots indicate high-risk proteins and green dots indicate low-risk proteins). (B) Overall survival analysis indicating that the prognosis of HNSCC in the high-risk group was better than that in the low-risk group. AXL: AXL receptor tyrosine kinase; BID: BH3 interacting domain death agonist; BRAF: B-Raf proto-oncogene serine/threoninekinase; CMET_pY1235: The MET transmembrane tyrosine kinase; CYCLIND1: Cyclin D1; E2F1: E2F transcription factor 1; ECADHERIN: E-cadherin; EEF2K: eukaryotic elongation factor 2 kinase; ERALPHA_pS118: estrogen receptor alpha; EZH2: enhancer of zeste homolog 2; HER3_pY1289: Erb-b2 receptor tyrosine kinase 3; HNSCC: head and neck squamous cell carcinoma; HR: hazard ratio; P27_pT198: cyclin-dependent kinase inhibitor p27; PAI1: plasminogen activator inhibitor type; PDCD4: programmed cell death 4; RAPTOR: mTOR regulation-related protein; SRC_pY416: SRC proto-oncogene.
Survival Results and Multivariate Examination
OS analysis showed that the expression of high-risk proteins was associated with better prognosis of HNSCC compared with low-risk proteins (P < 0.01; Fig. 1B). Also, the comprehensive prognostic signature model of the HNSCC revealed that HNSCC patients expressing high-risk proteins exhibited lower mortality rates and high OS (Fig. 2). Cox regression analysis showed that our prognostic model was an independent factor for OS (Fig. 3). The ROC curves were used to determine whether the expression pattern of survival-related proteins could provide an early prediction for the occurrence of HNSCC. We found an area under roc curve (AUC) of 0.720, which indicated that the sensitivity and specificity of this prognostic model were moderate. Clinicopathological factors associated with HNSCC were also explored. The AUC of age, gender, grade, stage, tumor (T), metastasis (M), and node (N) were 0.546, 0.476, 0.526, 0.639, 0.578, 0.444, and 0.698, respectively (Fig. 4).
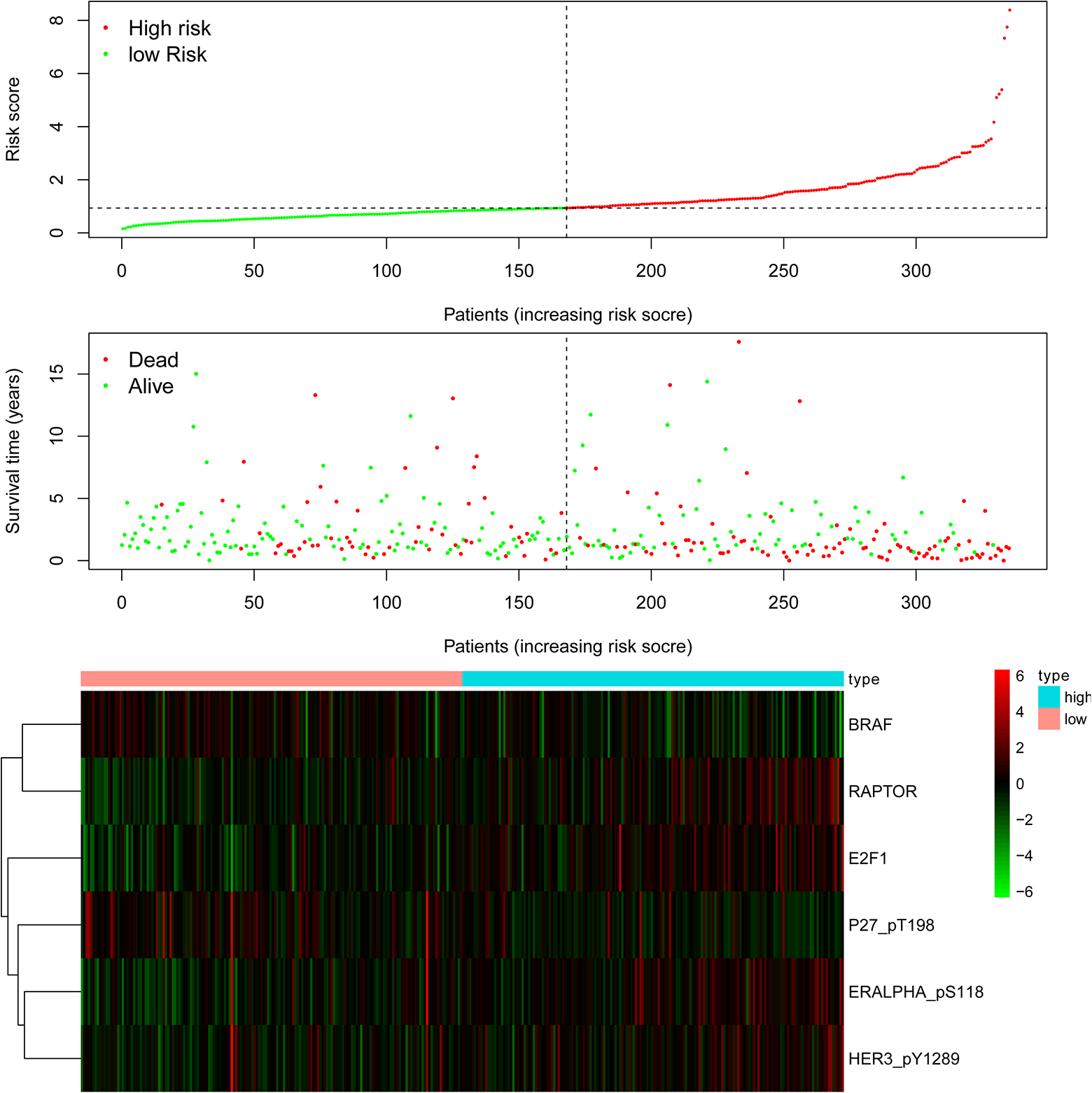
Detailed prognostic signature information of HNSCC groups. BRAF: B-Raf proto-oncogene serine/threoninekinase; E2F1: E2F transcription factor 1; ERALPHA_pS118: estrogen receptor alpha; HER3_pY1289: Erb-b2 receptor tyrosine kinase 3; HNSCC: head and neck squamous cell carcinoma; P27_pT198: cyclin-dependent kinase inhibitor p27; RAPTOR: mTOR regulation-related protein.
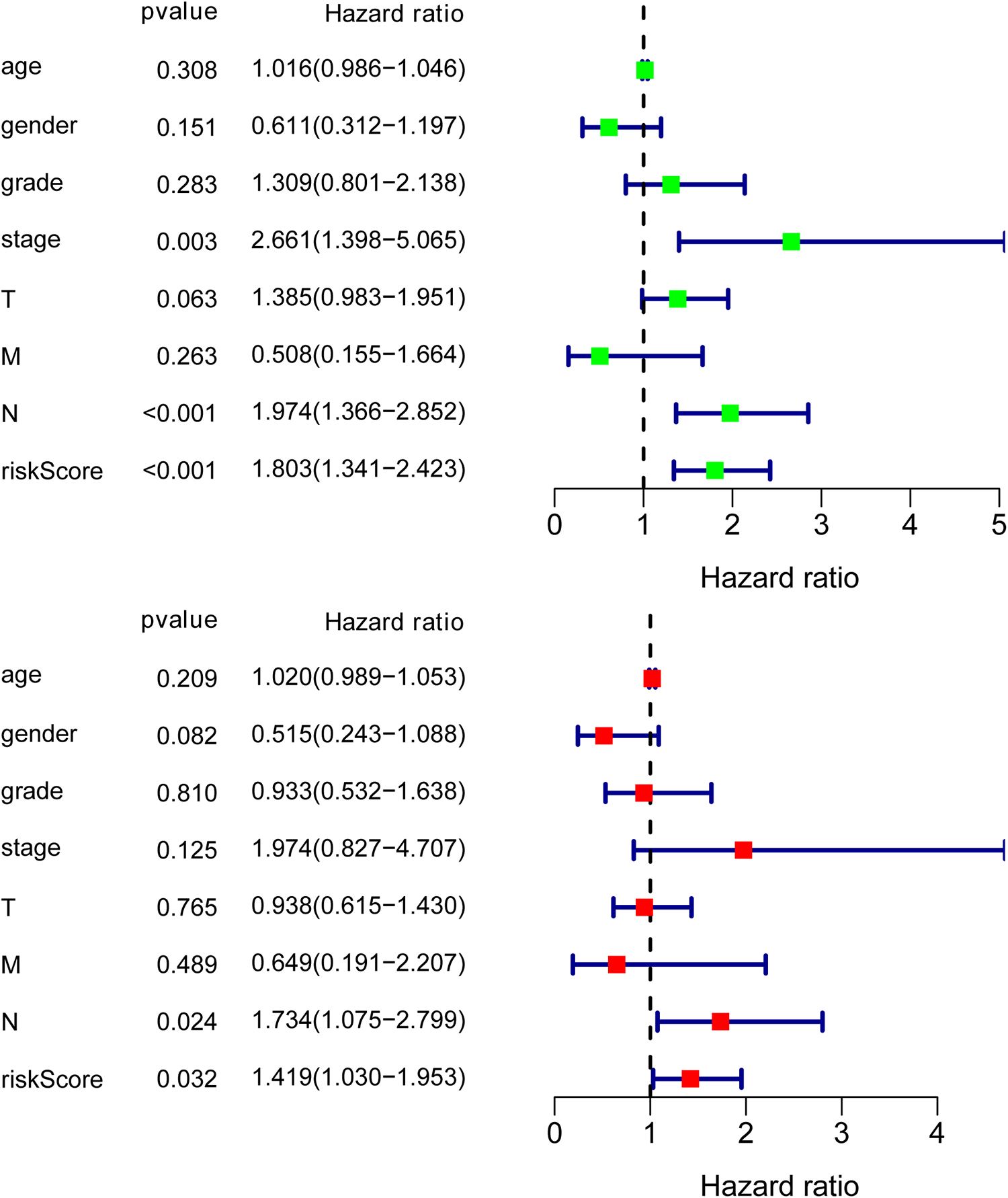
The results of Cox regression analysis indicate that our prognostic model is an independent prognostic factor for overall survival.
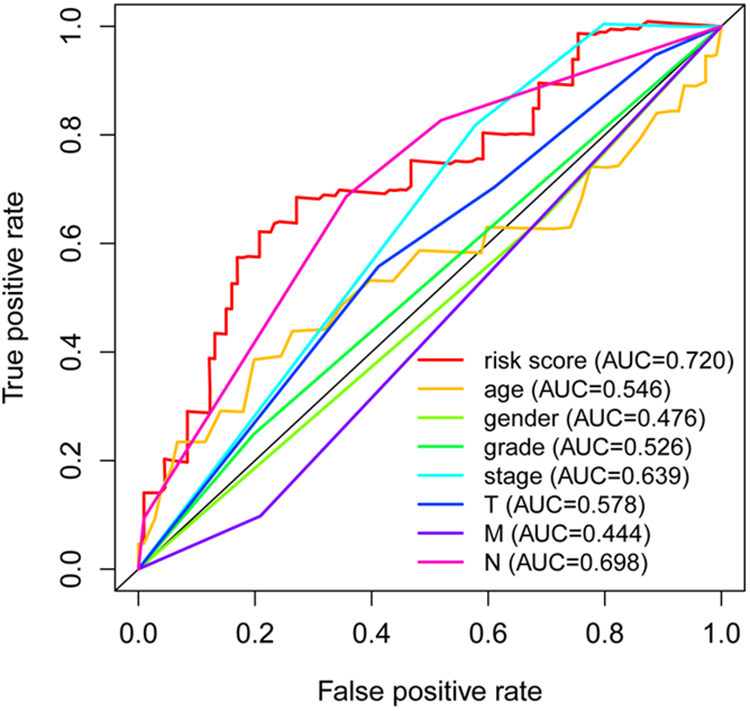
The results of our model and the clinicopathological predictors of HNSCC by ROC. AUC: area under roc curve; HNSCC: head and neck squamous cell carcinoma; ROC: receiver operating characteristic.
Creating Predictive Nomogram
A nomogram was created by combining clinical factors and the prognostic model. The final model comprised seven factors captured in TCGA, which included age, gender, grade, stage, T, M, and N (Fig. 5). Combining the prognostic model with clinical factors improved the accuracy of predicting OS for 1, 2, and 3 yr. Analysis of co-expression of the six proteins identified 24 negatively correlated and 68 positive correlated proteins (Table S2).

Nomogram constructed using the clinical pathology and prognosis models.
Discussion
Analysis of cancer proteomics provides new insights into changes that occur in the early stages of tumorigenesis, and therefore assist in the identification of novel biomarkers for early disease diagnosis and prognosis. Tumor cells present unique proteomic characteristics regarding their growth and survival compared to normal cells. These unique proteomic signaling pathways can be used as prognostic markers for the occurrence and development of cancer. Also, they can act as therapeutic targets in cancer treatment. Herein, we identified new and reliable prognostic proteomics signatures based on online datasets. These proteomic profiles are likely to manifest in patients with HNSCC and thus can be utilized as biomarkers and targets for therapeutic interventions.
In the present study, proteomic data was downloaded from TCPA (level 4), together with clinical survival data from TCGA. In total, 19 survival-related proteins were identified. The expression of E2F1 protein correlated with high OS. E2F1 (also known as E2F-1, RBAP1, RBBP3, RBP3) is a member of the E2F transcription factor family. The E2F family plays a vital role in controlling the cell cycle and inhibiting tumor-enhancing proteins. A recent study found that E2F1 activates lncRNA small nucleolar RNA host gene 3 (SNHG3), thereby promoting the migration and proliferation of cells in individuals with non-small-cell lung cancer by transforming the growth factor–β pathway 13 . Similarly, therapeutic targets that suppress the myeloid zinc finger 1 (MZF1) and MZF1 antisense RNA 1 (MZF1-AS1)/poly(ADP-ribose) polymerase 1 (PARP1)/E2F1 axis can inhibit the synthesis of proline and progression of neuroblastoma 14 . A study identified miRNA-205 as a putative HNSCC oncogene and regulator of E2F1 protein expression 15 . Notably, HNSCC caused by HPV infection expresses E6 and E7 viral oncogenes. These proteins suppress tumor protein p53 (TP53) and retinoblastoma 1 (RB1) and activate E2F1, a cell cycle regulator 16 . At present, the role of E2F1 in HNSCC has not been fully elucidated. Given this, we aimed to explore the relationship between the expression of E2F1 and the occurrence and progression of HNSCC. EZH2 and the catalytic component of Polycomb inhibition complex 2 are associated with the expression of homologous genes (Hox) and early stages of inactivating X chromosome 17 . Overexpression of EZH2 is thus associated with high incidence and poor prognosis of tumors. Maintaining the stability of mitochondrial membrane potential in HNSCC is dependent on EZH2 18 . Studies have shown that signal transducer and activator of transcription 3 (STAT3) signaling through overexpression/activation of phosphotidylinositol-3-kinase (PI3K) regulates HOX transcript antisense RNA (HOTAIR) and EZH2 in HNSCC, thus promoting the progression of the disease 19 .
The potential of utilizing the pattern of survival-related proteins in early prediction of future occurrence of HNSCC was examined using the ROC curve. The AUC was 0.720. We examined six proteins (ERALPHA, HER3, BRAF, P27, RAPTOR, and E2F1) in the multivariate Cox regression analysis to build the prognostic model. Estrogen receptor genes (ERalpha and ERbeta) play an important role in the progression of tumors, particularly breast cancer. Notably, the ERbeta gene has been shown to play a more significant role than ERalpha in the etiology of colorectal cancer 20 . In breast cancer, the expression of ERbeta alone is an independent risk factor for poor prognosis of patients undergoing tamoxifen adjuvant therapy 21 . The expression of ERalpha and ERbeta in HNSCC upregulates the estrogen receptor ligands, thus activating transcription and transduction of cytoplasmic signals 22 . Also, ERalpha was shown to be associated with epidermal growth factor receptor (EGFR) in HNSCC cells treated with dasatinib 23 . HER3 (Erb-b2 receptor tyrosine kinase 3) is a member of the human EGFR family and consists of four type-1 transmembrane tyrosine kinase receptors: HER1 to HER4. The upregulation of HER3 is associated with poor survival rates in various malignancies, including HNSCC and breast, colorectal, gastric, and prostate cancer 24 –26 . Studies have also found that upregulating HER3 using cetuximab hinders EGFR-mediated inhibition of HNSCC 27 . The small nuclear ribonucleoprotein polypeptide E (B-Raf) proto-oncogene serine/threonine kinase (BRAF) encodes a protein belonging to the serine/threonine-protein kinase family. Mutation in BRAF is a marker for response to respective targeted therapy in patients with HNSCC 28 . Therefore, factors that promote BRAF mutation drive oncogenesis. Accordingly, BRAF mutations are frequent in thyroid cancer 29 . P27 is a cyclin-dependent kinase inhibitor and plays an essential role in the negative regulation of the cell cycle. Overexpression of p27 inhibits the proliferation of cancer cells, increases apoptosis, and arrests cell cycle in HNSCC 30 . The expression of p27 can be used to predict how HNSCC responds to chemotherapy 31 . RAPTOR ((mechanistic target of rapamycin (mTOR) regulation-related protein) is a 150 kDa mTOR-binding protein that is essential for TOR signaling in vivo 32 . Activation of the serine-threonine protein kinase (Akt)-mTOR pathway is a common phenomenon in HPV-associated HNSCC. As such, mTOR inhibitors present attractive candidates for the treatment of HPV-positive squamous neoplastic lesions 33 . Similarly, activation of mTOR and auxiliary inhibition of mTOR in HPV-HNSCC disease can enhance primary site resistance, reduce the number of metastatic cells, and limit distant metastases 34 .
The signatures established in this study could reflect proteomic microenvironmental diseases and provide potential biomarkers for proteomic treatment and treatment response prediction. However, these signatures should be verified in more independent cohorts and functional experiments that predict proteins.
This study had two main limitations. First, the results were not validated using clinical samples, and second, the results may not be reliable because of the small sample size.
Conclusions
Based on the online database, we identified novel protein markers for the prognosis of HNSCC. The findings of the present study may provide new insights into the development of new and reliable tools for precise cancer treatment.
Supplemental Material
Supplemental Material, Sup - Data Mining Identifies Six Proteins that Can Act as Prognostic Markers for Head and Neck Squamous Cell Carcinoma
Supplemental Material, Sup for Data Mining Identifies Six Proteins that Can Act as Prognostic Markers for Head and Neck Squamous Cell Carcinoma by Zeng-hong Wu, Yun-Tang and Qing Cheng in Cell Transplantation
Footnotes
Authors’ Contributions
W.Z.H. designed and analyzed the research study; W.Z.H. and T.Y wrote and revised the manuscript, W.Z.H. and C.Q. collected the data; and all authors contributed to and approved the final version of the manuscript.
Availability of Data and Materials
Ethical Approval
As the work is a bioinformatics analysis article, ethical approval was not necessary and all the data were retrieved from the free online databases.
Statement of Human and Animals Rights
This article does not contain studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.