
Abstract
Sertoli cells (SCs) in the mammalian testes are well known as supporting cells of spermatogenesis, but have recently become an attractive source of cell therapy because of their capacity for immune modulation and trophic effects. In order to increase their applicable efficacy, we demonstrate a novel differentiation method for mouse embryonic stem cell (ESC)-derived Sertoli-like cells (SLCs) via the intermediate mesoderm (IM). We show that IM derived from an induction of 6 days expressed markers such as Wt1, Lhx1, Pax2 and Osr1, and that a sequential induction of 6 days resulted in ESC-SLCs. The SLCs expressed their marker genes (Sf1, Sox9, Gata4, Wt1, Fshr and Scf), but the pluripotency-marker gene Oct4 was decreased. After sorting by FSHR expression, high-purity (> 90%) SLCs were collected that showed distinct characteristics of SCs such as high phagocytic and immune modulation activities as well as the expression of immune-related genes. In addition, when transplanted into the seminiferous tubule of busulfan-treated mice, SLCs re-located and were maintained in the basal region of the tubule. These results demonstrated that our robust sequential differentiation system produced functional SLCs from mouse ESCs in vitro.
Introduction
Embryonic Sertoli cells (SCs) play a crucial role in the determination of the testis 1 . The testis-determining gene, Sry, is expressed between embryonic day (E) 10.5–12.5 in the male mouse gonad 2 and activates several other down-stream targets, including Sox9. Under this cascade of molecular signaling events, gonadal somatic cells can be differentiated into embryonic SCs 3,4 . Finally, the aggregation and rearrangement of the embryonic SCs and primordial germ cells (PGCs) result in the formation of testicular cords 1 . In the adult testis, SCs mainly regulate the differentiation of germ cells and support their maintenance inside the seminiferous tubules. In addition, SCs have immunosuppressive and trophic properties, which can be applied for the protection of non-testicular cells in transplantation procedures 5,6 . However, although SCs are considered a remarkable cell source for cell therapy, there are several drawbacks, such as the fact that mature SCs are quiescent somatic cells in the testis and have relatively lower proliferation activity when they resume proliferation in vitro 7 . Also, there is functional loss during primary culture of immature SCs 1 . These properties could limit the use of SCs for active applications. Therefore, investigating another source of SCs that could avoid these limitations may allow for their practical application.
Embryonic stem cells (ESCs), which are derived from the inner cell mass of blastocyst-stage embryos, have the unique ability to self-renew indefinitely during in vitro culture and have pluripotency, which permits their differentiation into all types of cells 8,9 . These capacities are spotlighted as an unlimited potential therapeutic source of cells for regenerative medicine 10,11 and can be a useful tool for the study of embryonic development 12 . ESCs exposed to the appropriate and specific conditions can be differentiated into cell types of all three germ layers (endoderm, ectoderm and mesoderm) and into germline cells 13 . During embryonic development of the mesodermal lineage, urogenital ridges are obtained via the intermediate mesoderm (IM) 14,15 . The urogenital ridge develops into three sets of tubular nephric structures (pronephros, mesonephros and metanephros), and the mesonephros among these tubular nephric structures is the central region from which the gonad arises 16 . Therefore, ESCs may be a main source for the robust large-scale production of gonadal somatic cells following the developmental process. In fact, it was recently shown that Sertoli-like cells (SLCs) can be derived spontaneously from human ESCs 17 , although the functional properties and purity of the differentiated SLCs remain unknown.
Therefore, the aim of this study was to establish a robust method of high-purity SLC production from ESCs. Our strategy was a three-step method following the developmental process, and the steps include the induction of the IM from mouse ESCs, the sequential induction into SLCs and sorting into high-purity SLCs using their specific markers. To the best of the authors’ knowledge, the present study is the first report to directly differentiate SLCs from mouse ESCs and to confirm their functional potential in vitro and in vivo.
Materials and Methods
Isolation of Mouse SCs
SCs for primary culture were obtained from the testes of 5-day-old and adult (>10-week-old) male C57BL/5 mice by a modified two-step enzymatic digestion protocol 18,19 . In brief, the tunica albuginea was removed from the testes manually, exposing the seminiferous tubules. The testes were then incubated in 1 ml DPBS containing 5 mg/ml collagenase IV (Gibco, Grand Island, NY), 0.1 mg/ml DNase (Sigma, St. Louis, MO) and 1 mg/ml Soybean trypsin inhibitor (Gibco) at 37°C for 10 min. After adding 2 ml DMEM/high glucose containing 10% FBS and sedimentation by centrifugation (×300 g for 5 min at room temperature (RT)), the supernatant was removed and the seminiferous tubules were incubated in 0.05% Trypsin-EDTA at 37°C for 5 min. The cells were washed three times with DPBS and centrifuged (×300 g for 5 min at RT). Following digestion, the cell suspension was filtered through a nylon mesh (Cell Strainer 100 μm; BD Falcon, Tokyo, Japan) to remove cell clumps and undigested materials. The filtrate was centrifuged and the supernatant was removed from the pellet. The cells in the pellet were then resuspended in complete culture medium, constituted of DMEM/high glucose medium supplemented with 10% FBS, 1% P/S, 1% NEAA, 0.1% β-mercaptoethanol. The cells were plated in a culture dish or in 6-well culture plates coated with 0.2% gelatin solution and incubated at 5% CO2 at 37°C in a humidified incubator. After culture for 2 days, the culture medium was changed to remove non-adherent cells from the dish or well. SCs from the testes of 5-day-old and adult mice were sampled for characterization (Fig. S1).
Maintenance of Mouse ESCs
The mouse ESC lines (karyotype: XY) were derived from a C57BL/6 strain mouse and from GFP-expressed transgenic mice [C57BL/6-Tg (CAG-EGFP), Japan SLC, Shuzuoka, Japan] and were cultured on irradiated mouse embryonic fibroblasts (MEFs; CF1 strain, Jackson Laboratory, Los GoTos, CA) as feeder cells. The mouse ESCs were maintained with ES cell culture medium consisting of 80% (v/v) DMEM high glucose (HyClone, Logan, UT) containing 20% (v/v), SR (Gibco-BRL, Frankin Lakes, NJ), 1% (v/v) NEAA (Gibco-BRL), 0.1% (v/v) β-mercaptoethanol (Gibco-BRL), 100 U/ml LIF (ESGRO, Chemicon, Temecula, CA) at 37°C in a 5% humidified CO2 incubator. For passaging, the mouse ESCs were detached from the dish by treatment with 0.05% trypsin-EDTA (TE; HyClone) for 3 min and were split onto a new MEF-seeded dish every 3–4 days. The mESC growth medium was changed daily.
Differentiation into Intermediate Mesoderm and then into Sertoli-Like Cells
Undifferentiated mouse ESCs were seeded at a density of 6×104 cells/cm2 onto Geltrex (Gibco-BRL)-coated plates in ES cell culture medium. At first, after an overnight culture, the cells were treated with Advanced RPMI (A-RPMI 1640; Gibco-BRL) supplemented with 100×L-GultaMAX (L-glu; Gibco-BRL), 1% penicillin/streptomycin (P/S; Gibco-BRL) and 5 μM CHIR99021 (Glycogen synthase kinase-3 inhibitor; Stemgent, Lexington, MA) for 36–48 h, followed by 100 ng/ml bFGF (Peprotech, Rocky Hill, NJ) and 1 μM retinoic acid (RA; Sigma) for 4 days to induce IM cells. The medium was changed after 2 days. For differentiation into SLCs, cells at the IM stage were treated with 100 ng/ml bFGF, 100 ng/ml FGF-9 (Peprotech), 500 ng/ml prostaglandin D2 (Santa Cruz Biotechnology, Dallas, TX), 10 ng/ml glia cell line-derived neurotrophic factor (GDNF; R&D, Minneapolis, MN), 10 ng/ml FSH (follicle stimulating hormone; Sigma) and 100×ITS (Insulin-Transferrin-Selenium; Invitrogen, Grand Island, NY) for 6 days. The medium was changed every 2 days.
Magnetic-Activated Cell Sorting (MACS) of SLCs Derived from Mouse ESCs
For isolating the mouse ESC-derived SLCs, FSHR, which is a testicular Sertoli cell marker, was used 20 . The differentiated cells (1×107) were trypsinized, collected, and were then incubated with anti-FSHR-biotin antibody (1:20, Bioss, Woburn, MA) for 30 min at RT in 100 μl of MACS solution (Miltenyi Biotec, Gladbach, Germany). Unbound anti-FSHR-biotin antibody was washed and removed by adding 1–2 ml of buffer and centrifuging at 300× g for 10 min two times. The cell pellet was resuspended in 80 μl of buffer, and then 20 μl of Anti-Biotin Microbeads UltraPure (Miltenyi Biotec) was added, mixed well, and incubated for 15 min at 4°C. The cells were then washed with 2 ml of 0.5% BSA (Sigma) in PBS buffer and centrifuged at 300×g for 10 min to remove the excess beads from the solution. Following disposal of the wash solution and according to the manufacturer’s guidelines for maximum column capacity, the pellet was resuspended with 500 μl of buffer, and the suspension was added to a prepped LD column (Miltenyi Biotec) fitted in a MACSMidi magnetic cell separator (Miltenyl Biotec). The column was washed with 2 ml of buffer two successive times to remove the unlabeled cells. Following column removal from the magnetic separator, the separated cells were eluted in 1 ml of buffer. The cell number was then determined.
Flow Cytometric Analysis
Pre- and post-sorted SLCs were collected and fixed with 4% paraformaldehyde for 10 min at RT. After washing, the cells were permeabilized with cooled 90% methanol for 10 min. Then, the cells were blocked with 0.5% bovine serum albumin (Sigma)/PBS for 30 min at RT and were incubated with primary antibodies. To evaluate the efficiency of FSHR MACS, the cells were incubated with an anti-rabbit FSHR antibody (Santa Cruz Biotechnology) and anti-mouse GATA4 antibody for 1 h at 4°C. Secondary antibodies were then detected using APC-conjugated (Life Technology, Rockford, IL) and PE-conjugated antibodies after an incubation for 1 h at 4°C. The control cells were not treated with primary antibodies. The cells were kept in the dark on ice until the analysis using a Becton DicKinson FACS IV Calibur (Becton Dickinson, San Jose, CA). At least 5,000 or 10,000 events were acquired for each sample.
Teratoma Formation
For the subcutaneous injection, mESCs or FSHR-positive SLCs were dissociated, mixed with 50 μl of Matrigel (BD Biosciences, San Jose, CA), and transplanted subcutaneously into the thigh of Balb/c nude mice. The mice received injections near the front legs. Teratoma formation was monitored over a period of 4–12 weeks. All mice received 1×106 cells per injection.
RNA Extraction, RT-PCR and Real-Time PCR
Total RNA was isolated from cells of each stage using TRIzol Reagent (Invitrogen) and was quantified using a NANODROP 2000 UV/Vis Spectrophotometer (Thermo Scientific, Waltham, MA). Reverse transcription (RT)-PCR was conducted using the First Strand cDNA Synthesis kit (Takara Bio, Shiga, Japan) and the AccuPower PCR premix (Bioneer, Deajeon, Korea) according to the manufacturer’s instructions. All PCR products were separated by 2% agarose gel electrophoresis. For quantification of expression levels, real-time PCR was performed with a final concentration of 25 ng of cDNA, using the iQ™ SYBR Green supermix (Bio-Rad Laboratories, Alfred Nobel Drive Hercules, CA) on the Bio-Rad CFX96™ Real-time PCR machine. The ΔΔCt method was applied to normalize the expression levels of each gene to those of Gapdh 21 . The following primers were used for RT-PCR and qPCR: Oct4 forward 5’-TGTGGACCTCAGGTTGGACT-3’; Oct4 reverse 5’-TTTCATGTCCTGGGACTCCTC-3’; Pax2 forward 5’-CTGTTTCCAGCGCCTCTAAC-3’; Pax2 reverse 5’-GACGCTCAAAGACTCGATCC-3’; Osr1 forward 5’-TTCGTTTGCAAGTTCTGTGG-3’; Osr1 reverse 5’-TGTAGCGTCTTGTGGACAGC-3’; Lhx1 forward 5’-CAGTGTCGCCAAAGAGAACA-3’; Lhx1 reverse 5’-TCAACGTCTCCAGTTGCTTG-3’; Wt1 forward 5’-CCAGTGTAAAACTTGTCAGCGA-3’; Wt1 reverse 5’-TGGGATGCTGGACTGTCT3’; Sox9 forward 5’-CACAAGAAAGACCACCCCGA-3’; Sox9 reverse 5’-GGACCCTGAGATTGCCCAGA-3’; Sf1 forward 5’-AGAAGTTTCTGAGAGCCCGC-3’; Sf1 reverse 5’-TACGAATAGTCCATGCCCGC-3’; Gata4 forward 5’-CTGGCCAGGACTGCCG-3’; Gata4 reverse 5’-GGTTGCTCCAGAAATCGTGC-3’; Fshr forward 5’- AATCCGTGGAGGTTTTCGCT-3’; Fshr reverse 5’-AGCACAAATCTCAGTTCAATGGC-3’; Scf forward 5’-GAAGACACAAACTTGGATTATCACT-3’; Scf reverse 5’-CATCCCGGCGACATAGTTGA-3’; TGF-β1 forward 5’-CCGCAACAACGCCATCTATG-3’; TGF-β1 reverse 5’-TGCCGTACAACTCCAGTGAC-3’; Transferrin forward 5’-TCTTCTCGGGCAGTTGTGTC-3’; Transferrin reverse 5’-CATGAGAAGGGATCCGAGCC-3’; Clusterin forward 5’-GGGTGTACTTGAGCAGAGC-3’; Clusterin reverse 5’-TCCTTGGAATCTGGAGTCCGGT-3’; Cyp11a1 forward 5’-TCCATTACCATCAGATGCAGA-3’; Cyp11a1 reverse 5’-GGGGTCCACGATGTAAACTG-3’; Hsd3b1 forward 5’-TGGACAAAGTATTCCGACCAG-3’; Hsd3b1 reverse 5’-TTCCAACACTGTCACCTTGG-3’; Hsd3b6 forward 5’-TGGACAAGTTCTTCAGACCAGA-3’; Hsd3b6 reverse 5’-TCTCCTTCCAACACTGTCACC-3’.
Immunofluorescence Analysis
The immunofluorescence analysis was performed as described previously 22 . Briefly, for immunocytochemistry, cells were fixed with 4% paraformaldehyde (PFA, Biosesang, Gyeonggi-do, Korea) in PBS for 1 h at 4°C and were permeabilized with 0.1% Triton X-100 (Sigma) in PBS for 5 min. Then, the cells were blocked with blocking solution (DAKO North America Inc., Carpinteria, CA) for 1 h at RT and were incubated with primary antibodies overnight at 4°C. Secondary antibodies were incubated for 1 h at RT. The following antibodies and dilutions were used: LHX1 (1:100; Santa Cruz); PAX2 (1:100; Thermo Scientific); GATA4 (1:100; Santa Cruz); and FSHR (1:100, Santa Cruz). The secondary antibodies were as follows: Alexa-488, Alexa-594, Alexa-488 (1:200, Life Technologies).
For immunohistochemistry, the primary antibodies used were as follows: GFP (1:100; Abcam, Boston, MA) and GATA4 (1:100; Santa Cruz). The secondary antibodies used were as follows: Alexa-488; and Alexa-594 (1:100, Life Technologies). Immunofluorescence images were taken using a confocal microscope (Carl Zeiss LSM 880, Oberkochen, Germany).
Phagocytosis Assay
The phagocytosis activity of the adult SCs, 5-day-old SCs, induced SLCs, FSHR-positive SLCs, FSHR-negative SLCs and MEFs were determined by measuring the uptake of fluorescein-labeled Escherichia coli (E. coli) using a commercially available kit, according to the manufacturer’s instructions (Vybrant Phagocytosis Assay kit, Molecular Probes, Eugene, OR). Briefly, each cell was seeded on 4-well plates at a density of 2×105 cells/well in SLC differentiating medium at 37°C with 5% CO2. After 12 h, each cell was rinsed with PBS and was incubated with SLC differentiating medium containing fluorescein-labeled E. coli overnight. At the end of the incubation period, each cell was washed twice to remove the extracellular non-phagocytosed E. coli, and 0.5 ml of a solution containing 100 ng/ml of Hoechst (stain for cell nucleases; Thermo Scientific) in PBS was added to each well and incubated for 15 min at 37°C. Afterwards, each cell was washed twice with PBS. Each cell was then imaged using a confocal microscope (Carl Zeiss LSM 880). The phagocytic activity was measured as the GFP particle-involving cells per total cells 23 using ImageJ (NIH, Bethesda, MD) software.
T-Cell Proliferation Assay
A cell suspension of splenocytes from 6- to 8-week-old male mice was prepared, and the erythrocytes were lysed with ACK (R7757, Sigma-Aldrich). The CD4+ T cells were purified by using a CD4+ T-cell Isolation kit (130-104-454, Miltenyi Biotech) according to the manufacturer’s instructions. The purity of the CD4+ T cells was confirmed using a CD4 antibody (FITC-conjugated anti-mouse CD4, 553729, BD Biosciences) and was greater than 96%. For the T-cell proliferation assay, CD4+ T cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; 565082, BD Biosciences). Freshly purified T cells were labeled with 1.2 μM CFSE for 10 min at 37°C and were then washed three times in complete medium. CFSE-labeled CD4+ T cells were co-cultured with adult SCs, ESC-derived SLCs, human bone marrow-derived mesenchymal stem cells (BM-MSC, PT-2501, Lonza, Allendale, NJ), or isolated spleen cells at a 5:1 ratio. The cells were co-cultured at 37°C for 5 days in RPMI1640 supplemented with 10% FBS (all from Life Technologies), 100 units/ml penicillin and 100 μg/ml streptomycin (Hyclone), 50ng/ml phorbol myristate acetate (PMA, P8139, Sigma-Aldrich), and 1μM ionomycin (P9657, Sigma-Aldrich). After co-culture, the proliferation of the CD4+ T cells was analyzed by assessing the intensity of the CFSE signal as determined via flow cytometry (Calibur, BD Biosciences).
Transplantation of SLCs into the Seminiferous Tubules of Busulfan-Treated Mice
The recipient animals (busulfan-treated ICR mice 24 ) were anesthetized using avertin, and the cell suspension (MACS-sorted cells [1×105 cells/testis; <10 μl]) was injected into the seminiferous tubules via the efferent duct 25 .
Statistical Analysis
All data presented are representative of at least three independent experiments unless otherwise indicated. The results are expressed as the mean ± SEM. Statistical significance was evaluated using one-way ANOVA followed by the Tukey test in the Statistical Package for the Social Sciences (SPSS, ver. 18; SPSS Inc., Chicago, IL). Statistical significance was defined as p < 0.05.
Results
Specification into the IM from Mouse ESCs
With the goal of establishing a differentiation procedure for mESCs into SLCs, we modified the method from Lam et al. (2014) 26 . Adhered ESCs were treated with 5 μM CHIR99021 for 36–48 h in order to induce the mesendoderm. The ESCs were then treated with 100 ng/ml bFGF and 1 μM RA for 4 days to generate the IM (Fig. 1A). After treatment with CHIR99021, the morphology of the cell clumps did not change. Furthermore, after treatment with bFGF and RA for 4 days, the adhered cells changed into a stretched shape and proliferated (Fig. 1B). During specification into the IM, the expression of IM gene markers (Pax2, Osr1, Lhx1 and Wt1) was determined by RT-PCR analysis. The expression of the IM markers was not detected in the cultured ESC-derived cells treated with CHIR99021 for 2 days. However, the expression of Pax2, Lhx1 and Wt1, but not Osr1, was initiated in those cells after treatment with bFGF and RA for 2 days. After treatment with bFGF and RA for 4 days, all IM markers were expressed in those cells (Fig. 1C), and their expression was confirmed by immunocytochemistry (Fig. 1D). These results indicated that ESC-derived cells in this condition were most likely to be representative of the IM.

Induction of mouse embryonic stem cells (mESCs) into intermediate mesoderm (IM). (A) Diagram showing the stepwise induction of mESCs into IM. (B) Phase contrast image of CHIR-treated mESCs at 2 days and the sequential treatment with bFGF and RA at 4, 5 and 6 days. (C) Oct4, Pax2, Osr1, Lhx1 and Wt1 mRNA expression levels in the induced IM derived from mESCs. (D) Quantitative RT-PCR analysis of Oct4, Pax2, Osr1, Lhx1 and Wt1 expression in the induced IM derived from mESCs. The mRNA level of each gene was normalized to Gapdh and is expressed relative to mESCs (mean ± SEM). The different superscripts indicate p <0.05. (E) Immunofluorescence staining of the IM markers (PAX2 and LHX1) in the induced IM derived from mESCs. The scale bars represent 50 μm.
Differentiation into SLCs from the ESC-Derived IM
To differentiate into SLCs, the IM was treated with bFGF, FGF9, prostaglandin D2, FSH, and GDNF for 6 days (Fig. 2A). After induction for 6–7 days, the induced SLCs formed a cord-like structure similar to immature mouse SCs (Fig. 2B). To test whether SLCs easily form a tubule-like structure, we cultured immature SCs from 5-day-old-mice and ESC-derived SLCs on Matrigel, which allows for the formation of cell aggregations and 3D hollow cord-like structures after 48 h (Fig. 2C), as demonstrated in a previous report 27 . These 3D cord-like structures were hollow, similar to a tubule, and generated a highly regular hexagonal array in the Matrigel. When the cells were cultured on Matrigel in the SLC differentiation media, web-like structures formed, which was consistent with the initiation of tubulogenesis. The mRNA levels of SC markers, such as Wt1, Sox9, Sf1, Gata4, Fshr and Scf, were increased in the differentiated SLCs (Fig. 2D), and the expression levels of SC-specific markers (GATA4 and FSHR) were high, as shown by immunostaining (Fig. 2E). These results showed that the ESC-derived SLCs differentiated under our culture conditions were similar to immature SCs from 5-day-old-mice.

Induction of mouse embryonic stem cell (mESC)-derived intermediate mesoderm (IM) into Sertoli-like cells (SLCs). (A) Diagram showing the induction of mESC-derived IM into SLCs. (B) Phase contrast image of the induced SLCs derived from mESCs at 3 days (3D) and 7 days (7D) from 5-day-old mouse Sertoli cells (SCs). (C) Comparison between the induced SLCs and immature SCs in their capability to form cord-like structures and tubulogenesis when cultured with 10% FBS or differentiation medium on Matrigel for 28 h. (D) Oct4, Wt1, Sox9, Sf1, Gata4, Scf and Fshr mRNA expression levels in the induced SLCs derived from mESCs and immature SCs. (E) Quantitative RT-PCR analysis of Oct4, Wt1, Sox9, Sf1, Gata4, Scf and Fshr in the induced SLCs derived from mESCs and immature SCs. The mRNA level of each gene was normalized to Gapdh and is expressed relative to mESCs (mean ± SEM). The different superscripts indicate p <0.05. (F) Immunofluorescence staining of the SC markers (GATA4 and FSHR) in the induced SLCs derived from mESCs and immature SCs. The scale bars represent 50 μm.
Purification of the SLCs Derived from mESCs
A slightly high expression of Oct4 in the SLCs may suggest that undifferentiated ESCs or differentiating SLCs remained in the population of differentiated SLCs. To purify the differentiated SLCs from the cultured materials, we performed MACS using the FSHR antibody 20 . Immunostaining showed that the proportion of FSHR and GATA4-double-positive SLCs increased dramatically after sorting (Fig. 3A). In addition, the flow cytometric analysis confirmed that the proportion of FSHR-positive SLCs in the post-sorted group was significantly higher than that of the pre-sorted group (90.0 ± 5.9% vs. 15.4 ± 2.0%), and most cells were positive for FSHR and GATA4 (Fig. 3B). Furthermore, the mRNA expression levels of the SC markers (Wt1, Sox9, Gata4, Fshr) were much higher in the FSHR-positive SLCs than in the FSHR-negative SLCs (Fig. 3C and 3D). These results indicated that the FSHR-positive SLCs revealed features of more matured SCs.

Purification of the Sertoli-like cells (SLCs) derived from mouse embryonic stem cell (mESC)-derived intermediate mesoderm (IM). (A) Immunofluorescence staining of the SC markers (FSHR and GATA4). The left panel shows the pre-sorted induced SLCs by the FSHR antibody and the right panel shows the post-sorted induced SLCs. The scale bars represent 50 μm. (B) Flow cytometric analysis of FSHR pre- and post-sorted induced SLCs. (C) Gene expression levels of FSHR-positive and negative induced SLCs. Oct4, Wt1, Sox9, Gata4 and Fshr mRNA expression levels in the FSHR-positive and negative induced SLCs. (D) The mRNA level of each gene was normalized to Gapdh and is expressed relative to mESCs (mean ± SEM). The different letters indicate p <0.05.
Undifferentiated ESCs or differentiating SLCs raise the possibility of a harbored tumorigenic potential 13 . Importantly, unlike undifferentiated ESCs and differentiating SLCs, FSHR-positive SLCs did not produce tumors when injected into immunocompromised mice (data not shown). No tumors were detected in the FSHR-positive SLC-injected animals over the span of 12 weeks. In contrast, the injection of undifferentiated ESCs produced readily visible teratomas (data not shown). These results indicated that the FSHR-positive SLCs were differentiated SLCs with a non-tumorigenic potential.
Phagocytic Activity and Immunological Characterization of the ESC-SLCs
To analyze the function of the SCs, we evaluated the phagocytic activity of the SCs and SLCs in vitro. The phagocytic activity of the FSHR-positive SLCs was significantly higher than that of MEF cells (somatic control) and FSHR-negative SLCs but was similar to that of adult SCs (Fig. 4A and 4B) (p < 0.05).
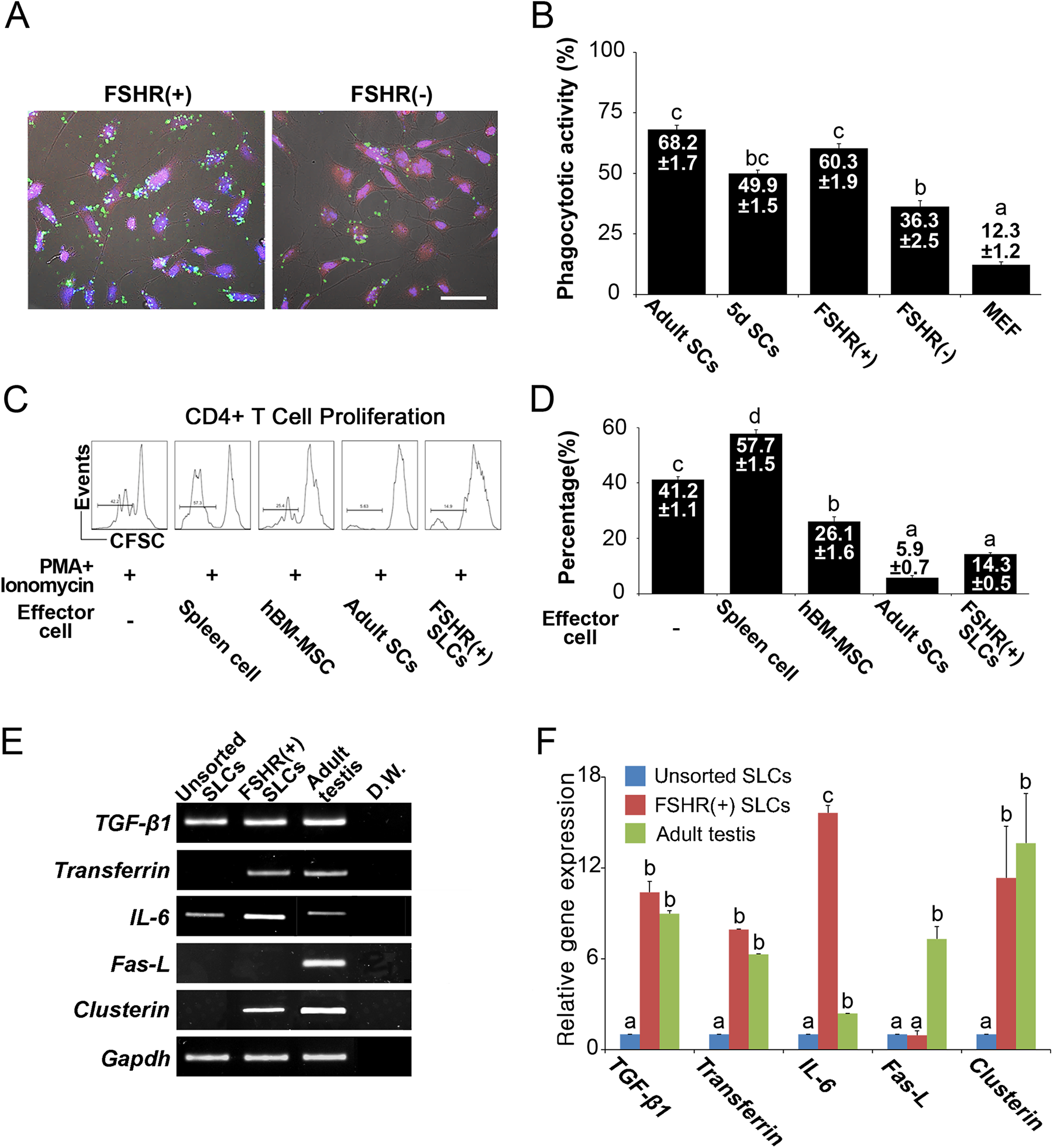
Sertoli-like cells (SLCs) have a high phagocytic activity and inhibit lymphocyte proliferation in vitro. (A) The phagocytic activity of the FSHR-positive SLCs and FSHR-negative SLCs. The left panel shows FSHR-positive SLCs. The right panel shows FSHR-negative SLCs. Blue, Hoechst-stained nuclei; Green, fluorescent microbeads; Red, induced SLCs derived from RFP-transfected mESCs. The scale bar is 40 μm. (B) Comparison of phagocytosis activity in various cells. The phagocytic activity was measured as the fluorescent microbead-involving cells/total cells (mean ± SEM). Different alphabet letters indicate p < 0.05. (C) Proliferation of CFSE-labeled CD4+ T cells co-cultured with SLCs under 50 ng/ml phorbol myristate acetate (PMA) and 1 μM ionomycin stimulations. PMA- and ionomycin-activated CD4+ T cells, syngeneic spleen cells, human BM-MSCs and adult Sertoli cells were used as controls. T-cell proliferation was measured after 5 days of co-culture. In the lower panel, the histogram summarizes the results shown in the upper panel. The data are shown as the mean (%) ± SEM. Values with different superscripts indicate a significant difference (p < 0.05). (D) The induced SLCs and FSHR-positive SLCs were subjected to RT-PCR analysis to assess the levels of the immunomodulation-related genes TGF-β1 Transferrin, IL-6, Fas-L and Clusterin. (E) Quantitative RT-PCR analysis of TGF-β1 Transferrin, IL-6, Fas-L and Clusterin in the induced SLCs and FSHR-positive SLCs. The mRNA level of each gene was normalized to Gapdh and is expressed relative to induced SLCs (mean ± SEM). The different superscripts indicate p <0.05.
During the co-culture, adult SCs and ES-derived SLCs significantly inhibited the proliferation of PMA-activated T cells, and the activity of immune modulation was higher than BM-MSCs (Fig. 4C). In addition, the mRNA levels of TGF-1 and Clusterin, both of which are immunomodulation-related genes, were highly expressed in the FSHR-positive SLCs and adult testis tissue. Furthermore, the mRNA of Transferrin, a marker of functional SCs, was detected in the FSHR-positive SLCs and adult testis tissue (Fig. 4D). These results indicated that FSHR-positive SLCs had an immunosuppressive function and features of matured SCs.
Integration of SLCs into the Seminiferous Tubule of Busulfan-Treated Mice
To determine the function of the ESC-derived SLCs, we next transplanted these cells into the testes of busulfan-treated recipient mice. For these experiments, EGFP-expressed SLCs were transplanted into the seminiferous tubules of busulfan-treated adult mice via the efferent duct. The SLCs in the recipient testes and seminiferous tubules expressed EGFP under UV light (Fig. 5A). By immunohistochemical analysis, small numbers of EGF-positive SLCs were observed in the basal area of the recipient seminiferous tubule and co-expressed GATA4, a SC marker, at 1 week post-transplantation (Fig. 5B). These results indicated that ESC-derived SLCs can be integrated into the seminiferous tubule of the recipient and may be a functional SC replacer.

Transplantation of FSHR-positive sorted Sertoli-like cells (SLCs) into the testes of busulfan-treated recipient mice, and the phagocytic activity of the various cells and expression of genes associated with immunomodulatory factors. (A) Busulfan-treated mouse testes of transplanted GFP-expressing, FSHR-positive sorted SLCs. The scale bar represents 5 cm. (B) Immunofluorescence staining using a GFP antibody for GFP-expressing FSHR-positive SLCs. The scale bar represents 20 μm. Anti-GFP (green) and anti-GATA4 (red) antibodies were used to stain the testis at 1 and 7 days post-transplantation. The upper panel demonstrates that the donor cells were detected in a cluster in the seminiferous tubule of the recipient testis at 1 day post-transplantation. The lower panel demonstrates that the donor cells were localized in the basal compartment of the recipient seminiferous tubule at 7 days post -transplantation. The scale bar represents 20 μm.
Discussion
Many studies suggest that testicular SCs may be a novel source of cells for cell therapy owing to their functional capabilities in immune modulation, anti-inflammation and nutritional support 1,19,28 . However, a lower mitotic activity of mature SCs is the main obstacle in securing a supply of cells. In the present study, we first applied a three-step method for differentiating SCs from mouse ESCs and thus obtained an unlimited source of SLCs with a high purity and the functions of testicular SCs.
A study on the differentiation of human ESC-derived SLCs was recently reported 17 in which a heterogeneous population of cells that contained at least three cell types, including PGCs, SLCs, and a small fraction of Leydig cells, was induced by spontaneous methods. In contrast to the previous report, we introduced a method for the directed differentiation into SCs based on the embryonic developmental process. At first, the stepwise differentiation system through the IM was applied in order to direct the cells into gonadal or renal lineages 15 . As shown in a recent report examining human ESC and induced pluripotent stem cells (iPSCs) 26 , treatment with CHIR99021 and sequential treatment with bFGF and RA in mouse ESCs induced IM cells (Fig. 1). In fact, this method was a rapid, efficient and highly reproducible system to induce IM cells, expressing the markers Wt1, Lhx1, Pax2 and Osr1, from pluripotent stem cells under our monolayer culture conditions. Then, the SLCs from the IM were induced by treatment with cocktails containing growth factors and FSH (Fig. 2), and their functions were well characterized in vitro and in vivo. Until now, a cell line with all the features of SCs has not been established 29 . In fact, immature and mature SCs, as well as established cell lines, lose their characteristics during prolonged culture 1 . Therefore, the establishment of ESC-SLCs with full characteristics may provide not only a source of cells for cell therapy, but also a research model for the study of SC functions. In the present study, the ESC-SLCs produced in vitro exhibited the appropriate characteristics and functions in vitro and in vivo.
Recently, other approaches for the induction of SCs were suggested. The in vitro generation of SLCs from human umbilical cord perivascular cells was reported by applying 5-step differentiation protocol that mimics the physiological phases of gonadal development 6 . In addition, more aggressively, Buganim et al. reported the direct reprogramming of mouse fibroblasts into embryonic SLCs using five transcription factors (Nr5a1, Wt1, Dmrt1, Gata4 and Sox9) in the mouse 29 . Although direct transdifferentiated SLCs from fibroblasts show similar functions as our ESC-SLCs (e.g., the formation of cord-like structures), the application of this method has several drawbacks, such as requiring much time, high cost and genetic modification.
In the testis, the maintenance of spermatogenesis inside the seminiferous tubules requires constant and intimate interactions between functional SCs and all stages of the differentiating germ cells. In fact, the main role of SCs is to provide support and nutrition to the developing sperm cells 29 . In addition, isolated SCs protect and facilitate the survival of non-testicular cellular grafts by their trophic factors 5,30 , as well as support germ cell differentiation by co-culture 1 . Thus, it was suggested that the establishment of a stable SC line from pluripotent stem cells can potentially support longer co-cultures of SC-germ cells, allowing for a more complete support of germ cells for infertility treatments. In addition, the robust production of ESC-SCs may allow for a novel utilization of stem cells in infertility treatments and cell therapies.
Conclusions
In this study, we established a novel differentiation method for mESC-derived SLCs through the IM. The SLCs were sorted by FSHR and showed distinct features of SCs, such as a high phagocytic and immune modulation.
Supplemental Material
Supplemental Material, mouse_sertoli-CT-2034-R1-Supplemental_data - In Vitro Derivation of Functional Sertoli-Like Cells from Mouse Embryonic Stem Cells
Supplemental Material, mouse_sertoli-CT-2034-R1-Supplemental_data for In Vitro Derivation of Functional Sertoli-Like Cells from Mouse Embryonic Stem Cells by Dong-Won Seol, Seah Park, Eun Young Shin, Jae Ho Chang, and Dong Ryul Lee in Cell Transplantation
Footnotes
Ethical Approval
Animals were used. The protocols for the use of animals in these studies were approved by the Institutional Animal Care and Use Committee (IACUC170068) of CHA University.
Statement of Human and Animal Rights
All of the experiments were carried out in accordance with the approved protocols of Institutional Animal Care and Use Committee (IACUC170068) of CHA University.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported partly by grants from the Bio & Medical Technology Development Program (2015M3A9C6028961 and 2017M3A9C8029318) of NRF and MSIP of Republic of Korea.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.