
Abstract
β-Cell replacement therapy via islet transplantation is a promising possibility for the optimal treatment of type 1 diabetes. However, such an approach is severely limited by the shortage of donor organs. Pancreatic stem/progenitor cells could become a useful target for β-cell replacement therapy in diabetic patients because the cells are abundantly available in the pancreas of these patients and in donor organs. In this study, we established a mouse pancreatic stem cell line without genetic manipulation. The duct-rich population after islet isolation was inoculated into 96-well plates in limiting dilution. From over 200 clones, 15 clones were able to be cultured for over 3 months. The HN#13 cells, which had the highest expression of insulin mRNA after induction, expressed PDX-1 transcription factor, glucagon-like peptide-1 (GLP-1) receptor, and cytokeratin-19 (duct-like cells). These cells continue to divide actively beyond the population doubling level (PDL) of 300. Exendin-4 treatment and transduction of PDX-1 and NeuroD proteins by protein transduction technology in HN#13 cells induced insulin and pancreas-related gene expression. This cell line could be useful for analyzing pancreatic stem cell differentiation. Moreover, the isolation technique might be useful for identification and isolation of human pancreatic stem/progenitor cells.
Introduction
Diabetes is the most prevalent and serious metabolic disease, and the number of diabetic patients worldwide is increasing. The reduction of insulin biosynthesis in pancreatic β-cells is closely associated with the onset and progression of diabetes. Thus, it is important to search for ways to produce sufficient insulin-producing cells for transplantation in diabetes. Islet transplantation has had renewed interest due to the recent improved success of this procedure (7,8,11,17,18,21). However, an abundant source of tissue that satisfies the demand for β-cells has yet to be found. In order to make such a therapy available to more than a small percentage of patients with diabetes, new sources of insulin-producing cells must be identified. This problem could be overcome if it were possible to generate transplantable islets from stem cells. One attractive approach for the generation of β-cells involves the expansion and differentiation of adult human pancreatic stem/progenitor cells, which are closely related to the β-cell lineage. Several studies have suggested that adult β-cells might originate from duct or duct-associated cells (1,3,4,10,12,19,24). Ductal progenitor cells in the pancreas would become a particularly useful target for therapies that target β-cell replacement in diabetic patients (21,22) because duct cell types are abundantly available in the pancreata of these patients and in donor organs. However, the experimental utility of these cells has been hampered by their limited survival in culture and by heterogeneous cell populations.
In this study, we established a mouse pancreatic stem cell line without genetic manipulation. The clonal cell line obtained, HN#13, expresses the pancreatic and duodenal homeobox factor-1 (PDX-1), also known as IDX-1/STF-1/IPF1, one of the transcription factors of β-cell lineage. Induction therapy with exendin-4 and with PDX-1 and BETA2/NeuroD transcription factors using protein transduction technology (10,12,14–16) stimulated the expression of insulin mRNA in the cells. This cell line could be useful for analyzing the molecular mechanisms regulating pancreatic stem/progenitor cell differentiation. Moreover, the isolation technique might be useful for identification and isolation of human pancreatic stem/progenitor cells.
Materials and Methods
Isolation and Culture of Mouse Pancreatic Islets and Duct Cells
Islets and pancreatic stem cells were isolated from the pancreas of 8-week-old mice (CLEA Japan, Inc. Meguro, Tokyo). Mouse studies were approved by the review committee of Kyoto University Graduate School of Medicine and Nagoya University Graduate School of Medicine. For islet isolation, the common bile duct was cannulated and injected with 2 ml cold M199 medium containing 2 mg/ml collagenase (Roche Boehringer Mannheim, Indianapolis, IN) (14). The islets were separated on a density gradient, hand-picked under a dissecting microscope to ensure a pure islet preparation, and used immediately afterward.
Pancreatic stem cells (ductal cell morphology) were isolated using a modified islet isolation method. Previous study showed that, after purification on a Ficoll gradient, the top interface (1.062–1.096 density range) was 50–95% islet cells with varying amounts of duct and degranulated acinar tissue, the middle interface (1.096–1.11 density range) contained 1–15% islets, duct, and degranulated acini, and the pellet was mostly well granulated acinar tissue with less than 1% islets (1). Therefore, the cells in the top and middle layers were used in this study. After hand-picking islets from the top and middle layers under a dissecting microscope, the remaining cells were stained by dithizone and the remnant islets were deleted. The duct rich population after islet isolation was then cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS; BIO-WEST, Inc., Logan, UT, S1560 Lot. # SO5094S1560). After cells attached and spread, nonductal cells (fibroblast morphology) were removed mechanically with a rubber scrapper. The “duct-like” cells were then inoculated into 96-well plates, cloned by limiting dilution, and cultured in DMEM with 20% FBS.
Cell Induction and Differentiation
For inducing cell differentiation, the cells were cultured in DMEM with 10% FBS, 10 nM exendin-4, 10 mM nicotinamide, 10 ng/ml KGF, 100 nM PDX-1 protein, and 100 nM BETA2/NeuroD protein for 7–10 days. For PDX-1 and BETA2/NeuroD protein, the cDNAs were amplified by PCR using appropriate linker-primers and then subcloned into the NdeI and XhoI sites of pET21b(+) (Novagen, Madison, WI) using a ligation kit (TaKaRa, Tokyo, Japan). BL21 (DE3) cells containing the expression plasmids were grown at 37°C to an OD600 of 0.8. Isopropyl-β-D-thiogalactopyranoside was added to a final concentration of 0.1 mmol/L, and the cells were then incubated for 12 h at 24°C. Cells were sonicated and the supernatants were recovered and applied to a column of Ni-nitrilotriacetic acid agarose (Invitrogen, San Diego, CA).
Semiquantitative RT-PCR
Total RNA was extracted from cells using RNeasy Mini Kit (Qiagen, Tokyo, Japan). After quantifying RNA by spectrophotometry, 2.5 μg of RNA was heated at 85°C for 3 min and then reverse-transcribed into cDNA in a 25-μl solution containing 200 units of Superscript II RNase H-RT (Invitrogen), 50 ng random hexamers (Invitrogen), 160 μmol/L dNTP, and 10 nmol/L dithiothreitol. The reaction consisted of 10 min at 25°C, 60 min at 42°C, and 10 min at 95°C. Polymerization reactions were performed in a Perkin-Elmer 9700 Thermocycler with 3 μl cDNA (20 ng RNA equivalents), 160 μmol/L cold dNTPs, 10 pmol appropriate oligonucleotide primers, 1.5 mmol/L MgCl2, and 5 units Ampli Taq Gold DNA polymerase (Perkin-Elmer, Norwalk, CT). The oligonucleotide primers and cycle number used for semiquantitative PCR are shown in Table 1. The thermal cycle profile used a 10-min denaturing step at 94°C followed by amplification cycles (1 min denaturation at 94°C, 1 min annealing at 57°C, and 1 min extension at 72°C) with a final extension step of 10 min at 72°C. The steps taken to validate these measurements were previously reported (12).
List of Gene-Specific Primers
TaqMan Real-Time PCR
Quantification of insulin mRNA levels was carried using the TaqMan real-time PCR system according to the manufacturer's instructions (Applied Biosystems, Foster City, CA, USA). PCR was performed for 40 cycles including 2 min at 50°C and 10 min at 95°C as initial steps. In each cycle, denaturation was achieved for 15 s at 95°C and annealing/extension was achieved for 1 min at 60°C. PCR was carried out in 20 μl of solution using cDNAs synthesized from 1.11 ng of total RNA. Standard curves were obtained using cDNAs generated from total RNA isolated from primary mouse islets. For each sample, the expression of insulin was normalized by dividing by the β-actin expression level. Mouse insulin-2 and β-actin primers are commercially available (Assays-on-Demand Gene Expression Products; Applied Biosystems).
Senescence-Associated β-Galactosidase Staining
Senescence-associated (SA) β-galactosidase staining was performed as previously described (13). Briefly, HN#13 cells at PDL 50 and PDL 150 were washed in PBS, fixed in 0.25% glutaraldehyde for 10 min on ice, and incubated at 37°C with a fresh staining solution consisting of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal; 1 mg/ml), 40 mM citric acid/sodium phosphate (pH 6.0), 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 150 mM NaCl, and 2 mM MgCl2. Human umbilical vein endothelial cells (HUVECs; PDL 4 and PDL 50) were used as controls for the SA β-galactosidase staining.
Tumorigenic Assay
To examine the potential tumorigenicity of HN#13 cells at PDL 50 and PDL 150, 1 × 107 cells were inoculated into the right thighs of severe combined immunodeficiency (SCID) mice. As a positive control, we transplanted 1 × 107 HeLa cells into the left sides of the SCID mice.
Results
Isolation of Pancreatic Stem Cells
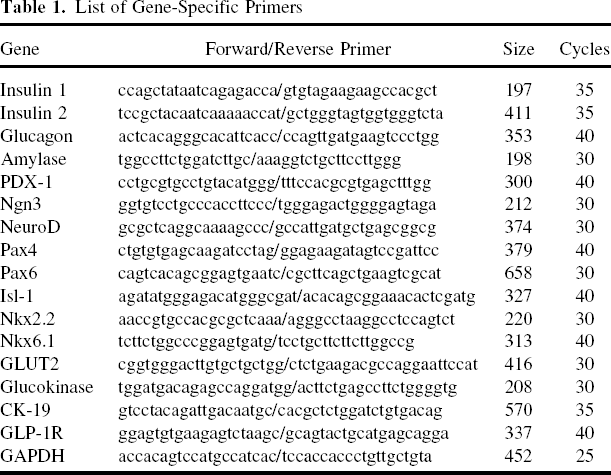
Pancreatic stem cells were isolated from the pancreata of 8-week-old mice. After hand-picking islets from the top and middle layers under a dissecting microscope, the duct rich population was then cultured in DMEM with 10% FBS. The “duct-like” cells were then inoculated into 96-well plates, cloned by limiting dilution, and cultured in DMEM with 20% FBS (see Materials and Methods). From over 200 clones, 15 clones were able to be cultured for over 3 months (Fig. 1). Clones #5, #12, and #13 formed a “cobblestone” morphology, whereas clones #2 and #11 formed a “fibroblast”-like morphology.

Isolation of pancreatic stem cells. Morphology of pancreatic stem cells. Pancreatic stem cells were isolated from the pancreas of 8-week-old mice. The duct rich population after islet isolation was cultured in DMEM with 10% FBS and nonductal cells were removed mechanically with a rubber scrapper. The “duct-like” cells were then inoculated into 96-well plates and cloned by limiting dilution. From over 200 clones, 15 clones were able to be cultured for over 3 months. Scale bar: 100 μm.
Selection of Pancreatic Stem Cell Line
All of these clones expressed PDX-1 and CK-19, markers for pancreatic ducts, but they did not express insulin mRNA (Fig. 2A). All of these clones also expressed glucagon-like peptide-1 (GLP-1) receptor (Fig. 2A) and the cells stimulated by induction medium including exendin-4 induced the expression of insulin mRNA (Fig. 2B). The clones (#5, #12, and #13) that formed the “cobblestone” morphology had higher expression of insulin mRNA than the other clones (Fig. 2B).

Selection of a pancreatic stem cell line. (A) Expressions of insulin, PDX-1, CK-19, and GLP-1 receptor in 15 clones. Fifteen clones were examined for the mRNA expression of insulin-1, insulin-2, PDX-1, CK-19, and GLP-1 receptor by RT-PCR. (B) Selection of a pancreatic stem cell line. For inducing cell differentiation, the cells were cultured in DMEM with 10% FBS, 10 nM exendin-4, 10 mM nicotinamide, 10 ng/ml KGF, 100 nM PDX-1 protein, and 100 nM BETA2/NeuroD protein for 7–10 days. The 15 clones stimulated by the induction medium were examined for the expression of insulin mRNA by real-time PCR.
Morphology and Growth Activity of the Pancreatic Stem Cell Line, HN#13
The clone #13 cells, named HN#13, had the highest expression of insulin mRNA after culture by induction medium. The cells formed a flat “cobblestone” monolayer that is characteristic of cultured duct cells (1). HN#13 cells continue to divide actively beyond the population doubling level (PDL) 300 (over 1 year) without changes in morphology (Fig. 3A) or growth activity (Fig. 3B).

Morphology and growth activity of pancreatic stem cell line, HN#13. (A) Morphology of the HN#13 cell line at PDL 50 (upper panel) and 150 (lower panel). HN#13 cells formed a flat “cobblestone” monolayer. Scale bar: 100 μm. (B) Growth activity of HN#13 cells. HN#13 cells both at PDL 50 and at PDL 150 could be maintained without growth inhibition.
FBS Dependency for Maintaining HN#13 Cell Line
The HN#13 cells were cultured in DMEM with 1 of 10 different lots of FBS. The cells cultured with only 1 of the 10 DMEM-FBS (BIO-WEST, Inc., Logan, UT, S1560 Lot. # SO5094S1560) formed the flat “cobblestone” monolayer. The cells in the other DMEM-FBS formed “fibroblast-like” or “spindle” structure and stopped dividing after three to six passages.
Senescence and Tumorigenicity of HN#13 Cells
Because HN#13 cells grew actively without change of morphology, the cells were tested to determine whether they entered senescence by using SA β-galactosidase staining. As shown in Figure 4A, neither PDL 50 (upper left) nor PDL 150 (upper right) cells showed marked SA β-galactosidase staining. By contrast, HUVECs entered senescence at PDL 50 (Fig. 4A, lower panels). To exclude the spontaneous transform of HN#13 cells, the tumorigenic potential in vivo was examined. HN#13 cells (1 × 107) at PDL 50 and PDL 150 were transplanted into SCID mice. No tumors developed in SCID mice receiving HN#13 cells at either stage during an observation period of at least 6 months (Fig. 4B). In contrast, sites injected with 1 × 107 HeLa cells developed tumors about 3 weeks after transplantation (Fig. 4B). These tumors were 40 mm in size 3 months later.
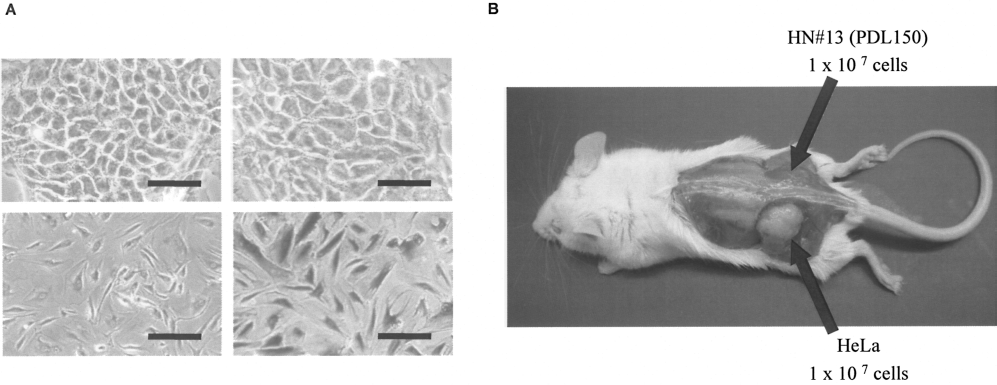
Senescence and tumorigenicity of HN#13 cells. (A) Senescence of HN#13 cell line. HN#13 cells were tested to determine whether they entered senescence using SA β-galactosidase staining. Upper left: HN#13 at PDL 50; upper right: HN#13 at PDL 150; lower left: HUVECs at PDL 4 (negative control); lower right: HUVECs at PDL 50 (positive control). Scale bar: 50 μm. (B) Tumorigenicity of HN#13 cells. To exclude the spontaneous transformation of HN#13 cells, the tumorigenic potential in vivo was examined. 1 × 107 HN#13 cells at PDL 150 and HeLa cells (positive control) were transplanted into SCID mice.
Cell Characterization Before and After Induction of Exendin-4 and Transduction of PDX-1 and BETA2/NeuroD Proteins
Although HN#13 cells expressed PDX-1 mRNA, which is one of the most crucial transcription factors for embryonic development of the mouse endocrine pancreas, insulin mRNA was not observed in HN#13 cells (Fig. 5A, B). On the other hand, other transcription factors such as Ngn3, BETA2/NeuroD, Pax4, Pax6, Isl-1, Nkx2.2, and Nkx6.1 were not expressed in untreated HN#13 cells. Transcripts encoding PDX-1, NeuroD, Pax4, Pax6, Isl-1, Nkx2.2, and Nkx6.1 were abundant in adult mouse islets. Neither glucagon mRNA nor amylase mRNA was observed in HN#13 cells (Fig. 5B).

Cell characterization before and after induction of exendin-4 and transduction of PDX-1 and BETA2/NeuroD proteins. (A) Insulin mRNA level of HN#13 cells before and after treatment with induction medium. HN#13 cells at PDL 50 and at PDL 150 were cultured in DMEM with 10% FBS, 10 nM exendin-4, 10 mM nicotinamide, 10 ng/ml KGF, 100 nM PDX-1 protein, and 100 nM BETA2/NeuroD protein for 7–10 days. The expression of insulin-2 mRNA was measured by real-time PCR. Mouse islets were used as a positive control. (B) Expression of other pancreas-related genes in HN#13 cells before and after treatment of induction medium. PCR was performed in a Perkin-Elmer 9700 Thermocycler with 2 μl cDNA (20 ng RNA equivalent) from human duct cells treated with adenoviruses. The oligonucleotide primers and cycle number used for semiquantitative PCR are shown in Table 1. Mouse islets were used as a positive control.
We previously reported that PDX-1 and BETA2/NeuroD protein transduction technology could be a safe and valuable strategy for facilitating the differentiation of stem/progenitor cells into insulin-producing cells without requiring gene transfer technology (10,12). The clone expressed GLP-1 receptor as shown in Figure 2A. Therefore, for inducing cell differentiation, the cells were cultured with exendin-4, PDX-1 protein, and BETA2/NeuroD protein for 7–10 days. The treated cells induced the expression of insulin mRNA, although the level of insulin mRNA was still low compared with primary mouse islets (Fig. 5A) and insulin in culture medium was undetectable (data not shown). We also examined the expression of other transcription factors and pancreas-related genes. After induction, Ngn3, BETA2/NeuroD, Pax4, Pax6, and Isl-1 transcription factors were induced, but not Nkx2.2, Nkx6.1, GLUT2, glucokinase, or amylase (Fig. 5B). These data suggest that HN#13 has a potential for differentiation into insulin-producing cells and the culture conditions induced insulin and pancreas-related gene expression in HN#13, although the induced cells are still immature.
Discussion
Replacement of the β-cell mass offers an alternative to standard insulin treatment and may overcome the long-term side effects associated with current therapies. This study demonstrates the isolation and the culture of adult mouse pancreatic stem cells that could be differentiated into insulin-producing cells in vitro. The ability to cultivate islets in vitro from digested pancreatic tissue that is usually discarded after islet isolation provides an important approach to β-cell replacement therapy. Our data provide evidence of the potential to expand and differentiate pancreatic ductal cells to islet cells, but further optimization of conditions is needed to generate a sufficient yield of islet tissue to make an impact on islet transplantation with glucose sensitivity.
HN#13 cells could be maintained by repeated passages for more than 1 year without growth inhibition. It is known that pancreatic stem/progenitor cells have a potential to differentiate into hepatocytes (23,24). We examined whether HN#13 cells can be differentiated into albumin-producing cells. For dexamethasone induction, HN#13 cells were differentiated into albumin-producing cells (unpublished data). Both pancreatic β-cells and hepatocytes are derived from the endoderm. We also examined whether HN#13 cells can be differentiated into adipocytes or osteoblasts, which are derived from the mesoderm. Although the induction methods for adipo/osteogenic differentiation are well established, the cells are rarely differentiated into adipocytes or osteoblasts (unpublished data). Therefore, HN#13 cells have the ability to differentiate into pancreatic β-cells and hepatocyte-like cells, but it is hard to get them to differentiate into mesodermal-derived cells, such as adipocytes or osteoblasts.
PDX-1, a homeodomain-containing transcription factor, plays a central role in regulating pancreatic development and insulin gene transcription. Furthermore, even in adults, PDX-1 is associated with islet neogenesis and the differentiation of insulin-producing cells from progenitor cells (1,5,20). We reported that PDX-1 protein can permeate cells by an Antennapedia-like protein transduction domain (PTD) sequence in its structure and that PDX-1, transduced into cultures of pancreatic ducts, induces insulin gene expression (12). BETA2/NeuroD, a basic helix–loop–helix (bHLH) transcription factor, is also a key regulator of pancreatic islet morphogenesis and insulin gene transcription (6,9). We reported that BETA2/NeuroD protein could be transduced into cells, due to an arginine- and lysine-rich PTD sequence in its structure (10). Transduced BETA2/NeuroD induces insulin gene expression. These data suggest that PDX-1 and BETA2/NeuroD protein transduction could be a safe and valuable strategy for enhancing insulin gene transcription and for facilitating the differentiation of ductal progenitor cells into insulin-producing cells without requiring gene transfer technology. GLP-1/exendin-4 has incretin effects, enhancing insulin secretion; it also stimulates β-cell replication and neogenesis and has antiapoptotic effects (2). Therefore, the HN#13 cells were cultured with exendin-4, PDX-1 protein, and BETA2/NeuroD protein for inducing cell differentiation. As shown in Figure 5A, the treated cells induced insulin mRNA. However, the level of insulin mRNA was still low compared with primary mouse islets, suggesting that the induced cells were still immature. Therefore, it is important to search for other ways to differentiate stem/progenitor cells into insulin-producing cells with the purpose of producing sufficient cells for transplantation in diabetes.
In addition, this cell line does not have tumorigenic properties. This is important for clinical application because one of major concerns of stem cell therapy is transforming into cancer.
In conclusion, we established a mouse pancreatic stem cell line. The clonal cell line continued to divide actively beyond the PDL 300 and differentiated into insulin-producing cells by induction medium containing exendin-4, PDX-1, and BETA2/NeuroD protein. This cell line could be useful for examining pancreatic stem cell differentiation. Moreover, the isolation technique might be useful for identification and isolation of human pancreatic stem/progenitor cells. Our observations have indicated that the pancreatic stem/progenitor cells could be useful for exploring their potential as a source for new β-cells.
Footnotes
Acknowledgments
The authors wish to thank Dr. Bashoo Naziruddin, Dr. Nicholas Onaca, Mr. Andrew Jackson, and Ms. Yoshiko Tamura (Baylor Research Institute) for technical advice, Dr. Carson Harrod for his careful reading and editing of this manuscript, and Ms. Nobuyo Hatanaka (The University of Tokyo) for assistance. This work was supported in part by the Juvenile Diabetes Research Foundation International (JDRFI); the Ministry of Education, Science and Culture, the Ministry of Health, Labour and Welfare; and Baylor All Saints Health Foundation.