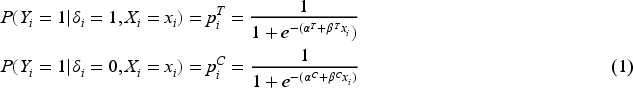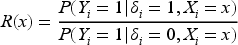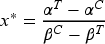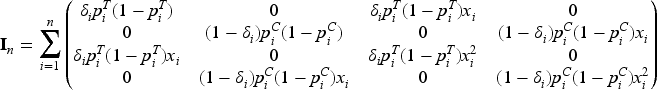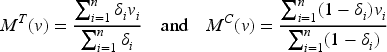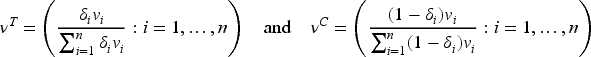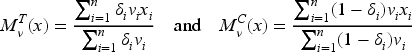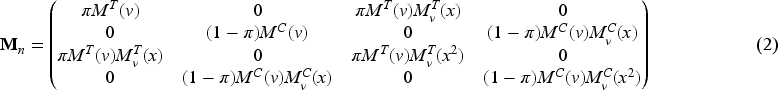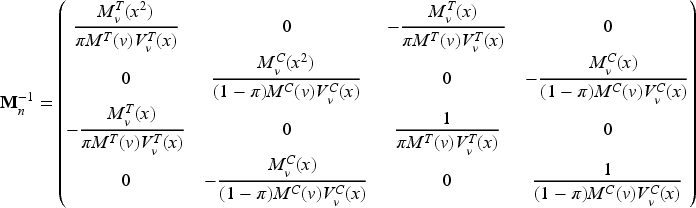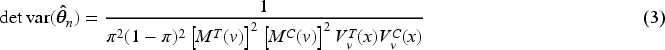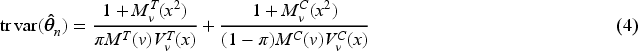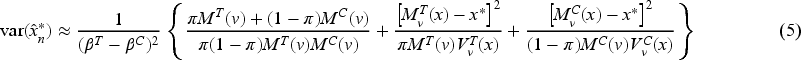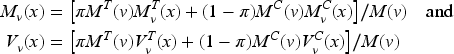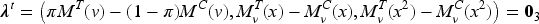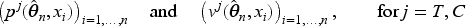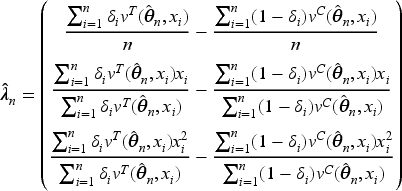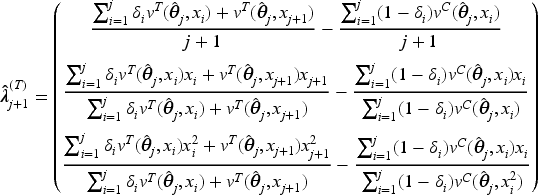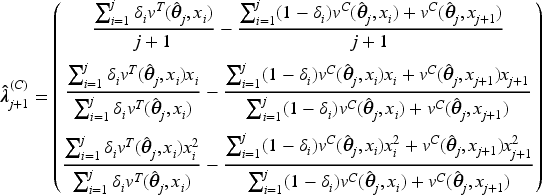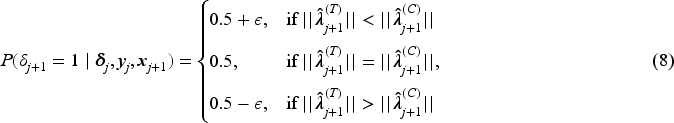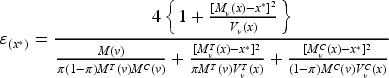The rapidly developing field of personalized medicine is giving the opportunity to treat patients with a specific regimen according to their individual demographic, biological, or genomic characteristics, known also as biomarkers. While binary biomarkers simplify subgroup selection, challenges arise in the presence of continuous ones, which are often categorized based on data-driven quantiles. In the context of binary response trials for treatment comparisons, this paper proposes a method for determining the optimal cutoff of a continuous predictive biomarker to discriminate between sensitive and insensitive patients, based on their relative risk. We derived the optimal design to estimate such a cutoff, which requires a set of equality constraints that involve the unknown model parameters and the patients’ biomarker values and are not directly attainable. To implement the optimal design, a novel covariate-adjusted response-adaptive randomization is introduced, aimed at sequentially minimizing the Euclidean distance between the current allocation and the optimum. An extensive simulation study shows the performance of the proposed approach in terms of estimation efficiency and variance of the estimated cutoff. Finally, we show the potential severe ethical impact of adopting the data-dependent median to identify the subpopulations.
Nowadays almost all branches of medicine are moving toward personalized medicine based on the belief that all patients cannot be successfully treated with the same therapy. In the presence of some evidence that the effect of a treatment may differ in certain subpopulations, personalized (also known as precision medicine) may be extremely useful. More precisely, personalized medicine is the tailoring of medical treatments to the individual characteristics, or biomarkers, of each patient. The process of selection of the subpopulations based on one or more biomarkers is called enrichment: subjects are screened for their biomarker profile, and then only those with or without certain characteristics are included in the trial and could be suitably randomized to the competing treatments.
While enrichment trials are specifically applied in the design phase, in terms of both enrollment restrictions and treatment allocation process, traditional subgroup identification methods are ex-post procedures for analyzing data already accrued in randomized clinical trials. The vast majority of them are classification algorithms of heuristic nature, without model specifications; they are based on classification trees combined with machine learning techniques, aimed at identifying a set of predictive biomarkers (among many available covariates, some of which are of prognostic nature) as well as suitable subsets of their support (see, e.g. Foster et al.1). Other proposals have been introduced for continuous endpoints under the classical linear model, often in the causal inference framework or inspired by latent variables approaches (see for a recent review by Loh et al.2).
In several cases, the effects of biomarkers on treatments are not explicit. Therefore, adaptive enrichment design methodologies, using the accrued information on previous subjects’ responses to find the benefitting population, can be applied. In the presence of a binary (or categorical) biomarker, the patients’ subgroups are well-defined. Nevertheless, when the biomarker of interest is defined on a continuous scale (e.g. age, cholesterol, and blood pressure), a widely used approach consists of discretizing the biomarker via a cutoff given by a data-driven quantile, usually the median, in order to define the subgroups. However, as stated by many authors, “discretization of a continuous biomarker using sample percentiles results in significant information loss and should be avoided” (see Polley and Dignam3 and Zhang and Molinaro4). Moreover, often a single candidate predictive biomarker is identified via preliminary information, but a suitable cutpoint has not been established.5 Under this framework, patient enrollment restriction based on the biomarker would be inappropriate. Indeed, an erroneous identification of the threshold for guiding future treatment decisions for individual patients could lead to potentially severe consequences.
Regrettably, there is still a lack of studies that estimate the threshold directly on a continuous scale to correctly discriminate among the so-called sensitive (biomarker-positive) and insensitive (biomarker-negative) patients.6 Recently, for a single-arm trial, Spencer et al.7 presented a biomarker-adaptive threshold design to determine if a subpopulation with a clinically relevant response rate exists. Some proposals have been developed in the context of survival trials (see Trippa et al.,8 Renfro et al.,9 and Diao et al.10). On the other hand, Lin et al.11 and Frieri et al.12 adopted a bivariate-normal model to estimate the biomarker threshold, using the correlation biomarker-response as an explicit gauge of the predictive nature of the biomarker. Baldi Antognini et al.13 used the linear treatment-by-covariate interaction approach to find the cutoff and provide the optimal allocations for both parameter estimation and cutoff identification.
Inspired by numerous real clinical trials documented in the literature, consider as a motivating example the NSABP B-35 trial described by Margolese et al.14 By enrolling 3104 patients, this study compares anastrozole versus tamoxifen in postmenopausal women with hormone receptor-positive ductal carcinoma in situ undergoing lumpectomy plus radiotherapy, showing that anastrozole provides a significant improvement in breast cancer-free interval in women younger than 60 years.
Since a binary model for the biomarker–treatment relationship is frequently required for therapeutic success/failure cases (see, e.g. Vinnat and Chevret15), in this paper, we approach the challenging issue of identifying the optimal cutpoint of a continuous predictive biomarker based on the relative risk for binary trials for treatment comparisons. Our aim is to provide optimal allocations for inference on the threshold and then describe a suitable covariate-adjusted response-adaptive (CARA) procedure to implement this optimum. After introducing the statistical model in Section 2, Section 3 deals with classical and optimality criteria, also providing the variance of the estimated threshold. We derive the optimal design that simultaneously optimizes all the above-mentioned criteria, which consists of a set of conditions, involving the unknown patients’ biomarker values and the unknown model parameters. Thus, Section 4 is dedicated to the implementation of the optimal design through a suitably defined CARA rule. Finally, in Section 5, we perform a simulation study adopting normal and log-normal distribution for the biomarker, in order to compare the performance of the new CARA procedure with that of conventional trial designs. To stress the practical impact of our procedure, we redesign a clinical trial for severe sepsis disease, taking also into account the Virtual Twins method1 for subgroup identification. Additionally, the ethical impact resulting from the use of a median-based biomarker cutoff has been explored, comparing it with the benefit gained from using the relative risk-based cutoff.
Binary responses
We consider an oncological trial where patients are sequentially assigned to one of two competing anticancer agents or . Let us denote by the chosen quantitative predictive biomarker, which is not under the experimenters’ control. When the th subject is ready to be randomized, the biomarker value is recorded and she/he receives one of the agents based on a given randomization rule and a treatment indicator variable records the assignment: if the patient is assigned to , while otherwise. Thus, is the subjects’ proportion allocated to and to . We assume independent and identically distributed random variables having common density function with finite expected value and variance. A binary tumor status is examined after the treatment assignment to directly measure any anticancer activity (e.g. the ctDNA clearance in Spreafico et al.16) and we take in the case of successful response of the th patient (clearly, this setting applies to any dichotomous outcomes of interest, such as mortality of patients with hypoxemic acute respiratory failure after different oxygenation therapies in the HIGH clinical trial, see Azoulay et al.17).
Conditionally on the treatments and the biomarker, subjects’ responses are assumed to be independent following a logistic regression model:
where, since is a predictive biomarker, .
The primary goal of the paper is to assess whether an anticancer agent is better than the other for a subgroup of subjects based on their biomarker. By taking into account the relative risk,
it is easy to show that, for every subject with biomarker value greater than the unique cutoff
agent is better than (namely ) provided that (or for if ). This subpopulation of patients (often referred to as target or benefitting subpopulation) is generally the focus of the investigator assuming that is regarded as the new/experimental treatment. Moreover, after the identification of the cutoff, a secondary objective is to evaluate the impact of the discretization of the continuous biomarker using a sample percentile instead of , in terms of ethical loss (namely the percentage of subjects assigned to the inferior treatment).
As regards the notation, throughout the paper let , , and be the vector of biomarker values, assignments, and outcomes observed after steps and is the -dimensional vector of zeros.
Trial designs
Optimal design theory is recognized as an important tool to achieve allocations that yield the best performance out of a range of potential designs. The literature is rich with optimal design methodologies that can be adopted by clinical researchers to improve the efficiency of the drug development process. The key idea is to select a criterion that measures the loss as a function of the inverse of the Fisher information matrix since it is an asymptotic approximation of the variance–covariance matrix of the maximum likelihood estimators (MLEs). Denoting by the vector of the unknown parameters, let be the MLE of after steps, then the Fisher information is (see Appendix 1)
To simplify the notation, let us define the variance of the response of patient with ; clearly, if then , whereas if . Thus, and , so that
simply denote the means of the observed variances in the two groups. Moreover, since
represent two discrete probability distributions, we denote by
the mean of the biomarker with respect to and , respectively, with () the corresponding variances. Thus, the average information matrix is
and its inverse is given by (see Appendix 2)
Optimality criteria
When the interest is in the estimation of the whole parameter vector , and optimality criteria are typically used. The former criterion minimizes the volume of the confidence ellipsoid for , so that the optimal design minimizes the determinant of . Whereas, the optimality minimizes the mean variance of the estimators, so the trace of should be minimized. Concerning the estimation of , the MLE of the threshold after steps is and the next theorem provides an approximated closed-form for its variance, as well as the expression of and optimality criteria.
After assignments,
and
As regards the threshold estimation, since , through a first-order approximation,
See Appendix 3.
Optimal designs
Assuming model (1), the following theorem provides the optimal design for the estimation of the cutoff, which is also and optimal.
An allocation satisfying
is and optimal and is optimal for estimating the threshold as well.
See Appendix 4.
Conditions (6) define a vast class of optimal designs through equality constraints, involving both the observed biomarker values and the unknown model parameters. In practice, optimality requires the equality of the biomarker weighted means and variances with respect to and , as well as the equality of to .
Since in our setting incoming patients’ biomarker cannot be controlled by the experimenter, given and the marginal quantities and (i.e. mean and variance evaluated w.r.t. ), the selection of and uniquely determines and :
Thus, conditions (6) can be restated as , , . If, in addition, a balanced allocation is assumed, the optimum requires and clearly and .
The dependence of the optimal designs (derived in Theorem 3.2) on the unknown model parameters is highlighted in the next remark.
Unlike the results obtained in Baldi Antognini et al.,13 conditions (6) cannot be achieved in practice, since they depend on all the observed allocations and biomarker values, as well as on the unknown model parameter . Thus, even in the unrealistic scenario in which the experimenter could be able to choose the biomarker values of the subjects, the closeness to the optimum cannot be checked since it depends on . Indeed, consider the following toy example with , where we assume and . In such a case , and , so the equality between the first and second empirical moments of the biomarker is achieved. When (i.e. ), conditions (6) are satisfied with , , and . However, if (inducing the same threshold ), then the considered allocation is not optimal since , , and . Moreover, under this choice of does not exist an allocation satisfying conditions (6).
CARA procedure to implement the optimum
As pointed out previously, the optimal designs derived in Theorem 3.2 depend on the patients’ biomarker values and the unknown model parameters. In these settings, CARA randomization could provide a possible solution to asymptotically approach the optimum. However, standard CARA procedures proposed in the literature are based on a target, namely on the complete specification of the functional form of the optimal design (see, e.g. Baldi Antognini and Giovagnoli18) and they are not applicable in this setting, where optimality is characterized only by equality constraints. Thus, we now propose a new CARA randomization procedure to achieve the optimum in a sequential manner. From (6), the optimal design requires that
so we suggest the biomarker-adjusted response-adaptive (BiomARA) to minimize the Euclidean norm sequentially. It goes without saying that depends, at each step , on the set of collected allocations and patients’ biomarker values, as well as on the unknown , namely , so it needs to be sequentially estimated taking also into account patients’ responses. To stress this dependence, from now on we let for every and , with . After assignments, we could evaluate the current MLE of and so estimate all the previous individual success probabilities and response variances, respectively, by
Thus, can be estimated via
and the BiomARA procedure can be implemented according to the following algorithm:
assign patients with restricted randomization to and and set ;
based on , and , estimate via ;
when the th patient arrives with biomarker value , calculate and in order to evaluate the potential distances and from the optimum that will occur by assigning or to this subject, where
if
if
assign the th patient to with probability
where , and then observe .
Set and repeat Steps 1–3 until the enrollment process is completed.
It is important to notice that, at Step 2, the response of the th patient is yet to be observed, so and depend on the biomarker value of patient but are calculated via .
Given the allocation function in (8), the experimenter is able to control the degree of randomness of the procedure through . At the extremes, the BiomARA with coincides with the completely randomized design, while for the procedure becomes deterministic.
Simulation results
To show the performances of the BiomARA, the present section is dedicated to its operating characteristics via an extensive simulation study. The suggested CARA procedure is implemented under several experimental scenarios corresponding to various hypothetical clinical settings, also in comparison with the permuted block design (PBD) having block size 4 and the complete randomization (CR), as standard benchmarks commonly used in practice. The patients’ biomarker has been simulated under two different distributions: a standard normal and, to take into account a nonsymmetrical distribution on , a log-normal (LN) distribution such that . For each scenario, we simulate 10,000 trials and we explore the behavior of BiomARA with and , denoted by BiomARA and BiomARA, respectively.
Efficiencies
To evaluate the relative efficiencies of a specific design, we consider and efficiency, denoted by and , whose analytical expressions are reported in Appendix B. These measures are used to compare a design with the optimum in terms of the corresponding criteria, with values equal to 1 in the case of maximum efficiency (see e.g. Atkinson et al.19). Analogously, a measure of efficiency in terms of threshold estimation could be defined as the ratio of the variance in (5) calculated in the optimum (6) and its value in a generic design, that is,
Adopting the optimal designs in (6) implemented via the BiomARA, we simulate the behavior of the above-mentioned efficiency measures, also including CR and PBD.
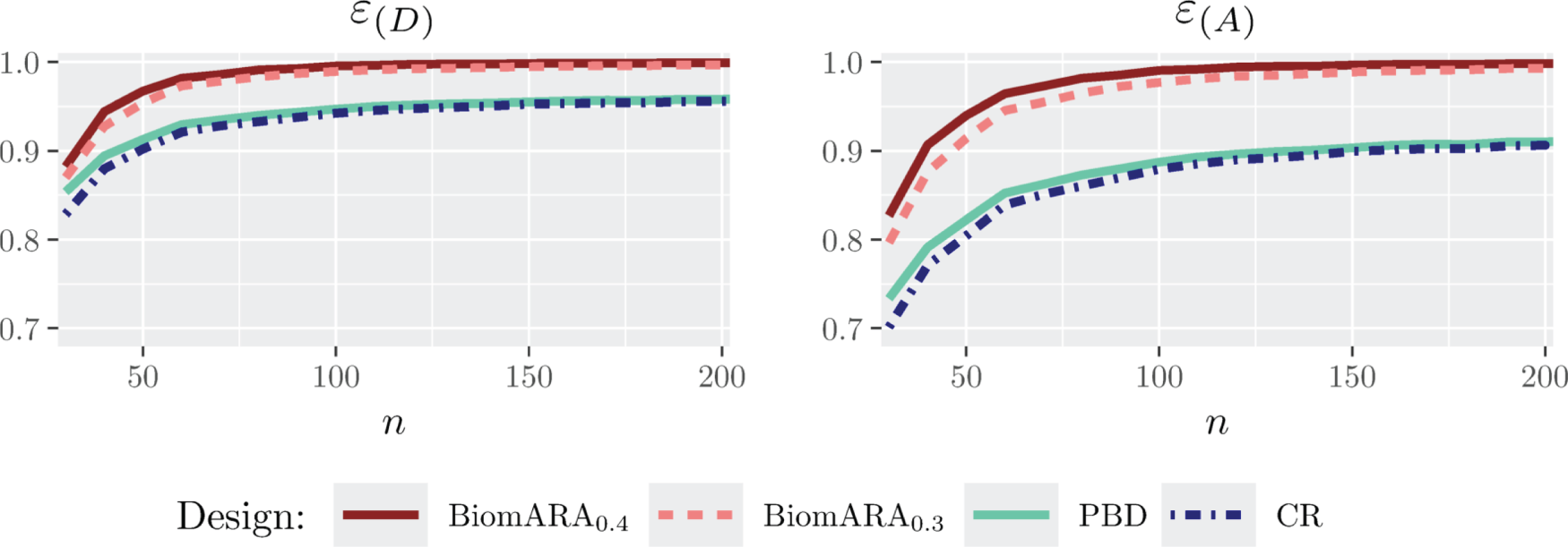
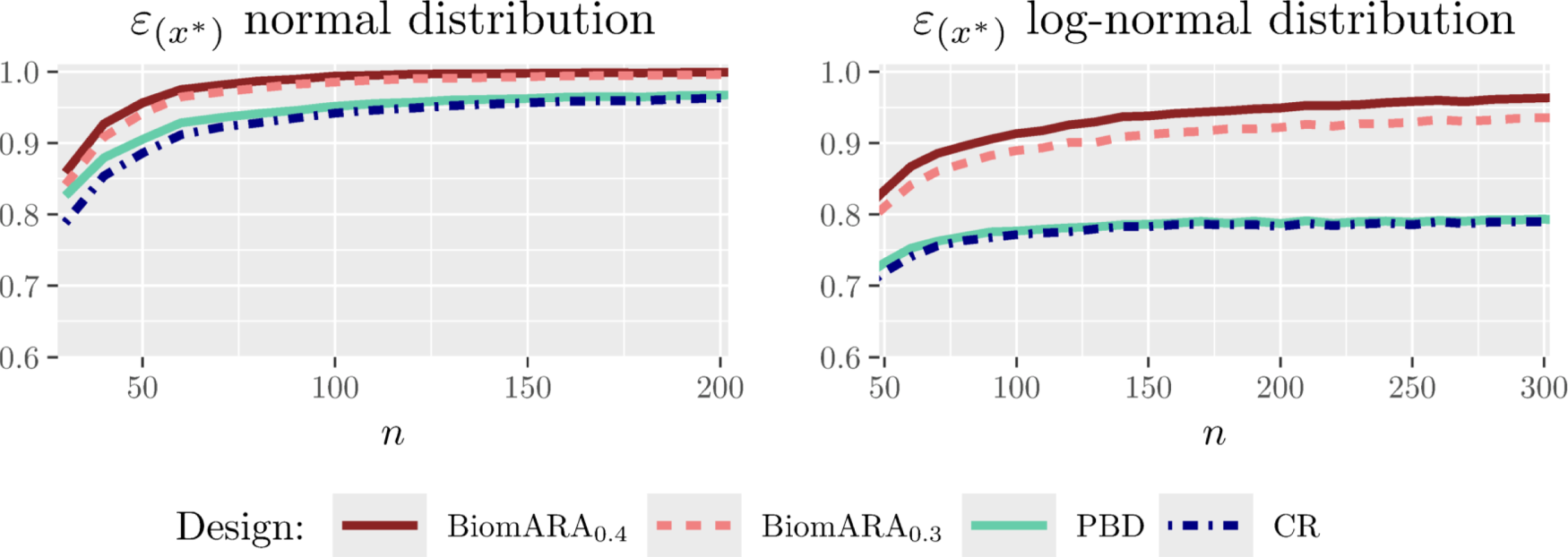
Figure 1 and the left panel of Figure 2 show the results for the standard normal distribution with as varies between and . Whereas, Figure 3 and the right panel of Figure 2 illustrate the results under the LN distribution with for between and . In both cases, BiomARA always shows a significant gain in terms of efficiency with respect to PBD and CR. Contrary to the behavior of PBD and CR, the efficiency under BiomARA increases as grows and, for the normal biomarker, reaches its maximum value even for very small sample sizes.
Since PBD and CR have similar performance in terms of all the measures of efficiency and in accordance with the recent literature concerning the selection of the biasing probability in the presence of covariates (see e.g. Ma and Hu20), in the following simulations we omit both CR and BiomARA.
and efficiency for standard normal biomarker distribution and .
Efficiency of threshold estimation when the biomarker has a standard normal distribution and (left panel) and when the biomarker has a log-normal distribution and (right panel).
and efficiency for log-normal biomarker distribution and .
Variance of the estimated cutoff
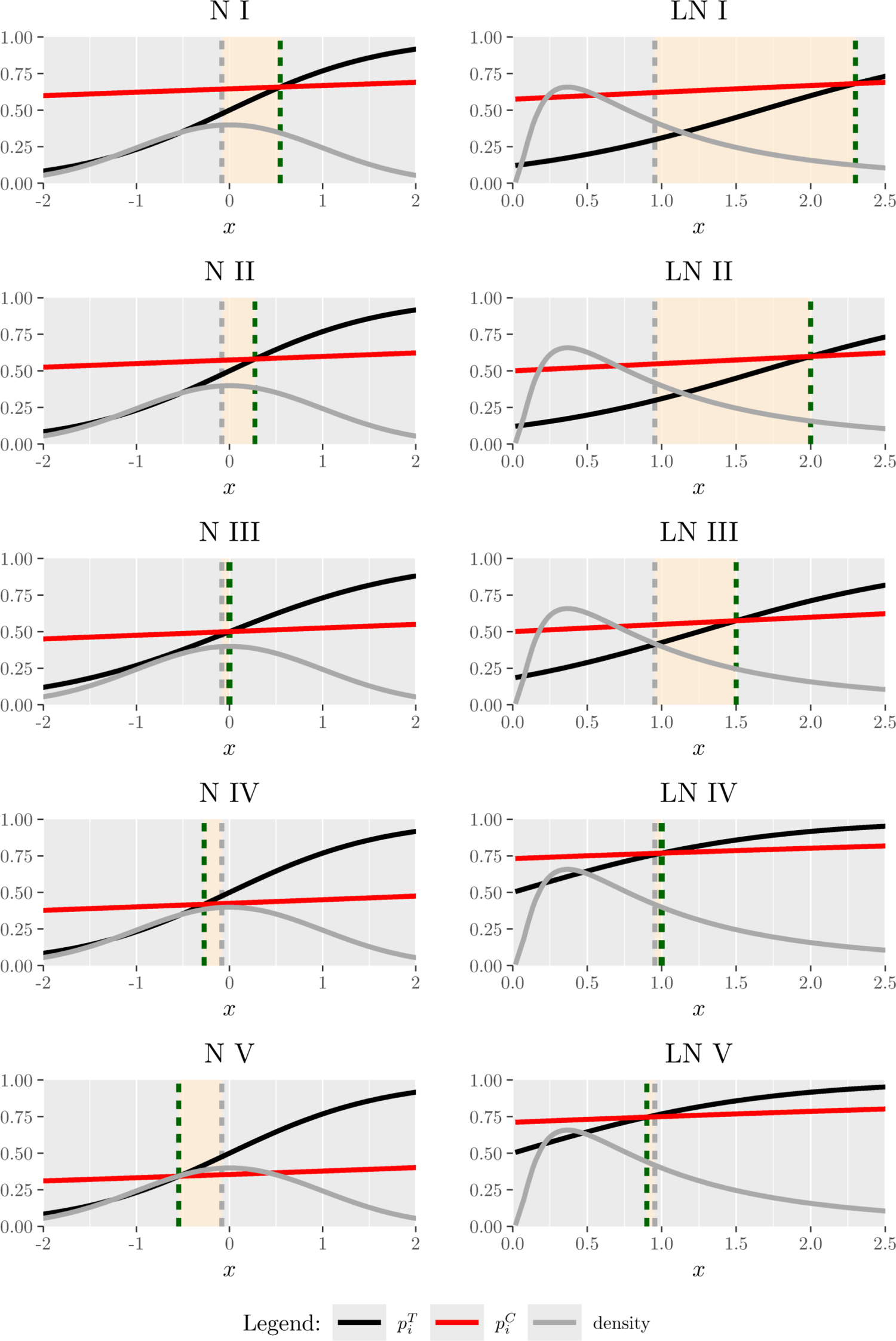
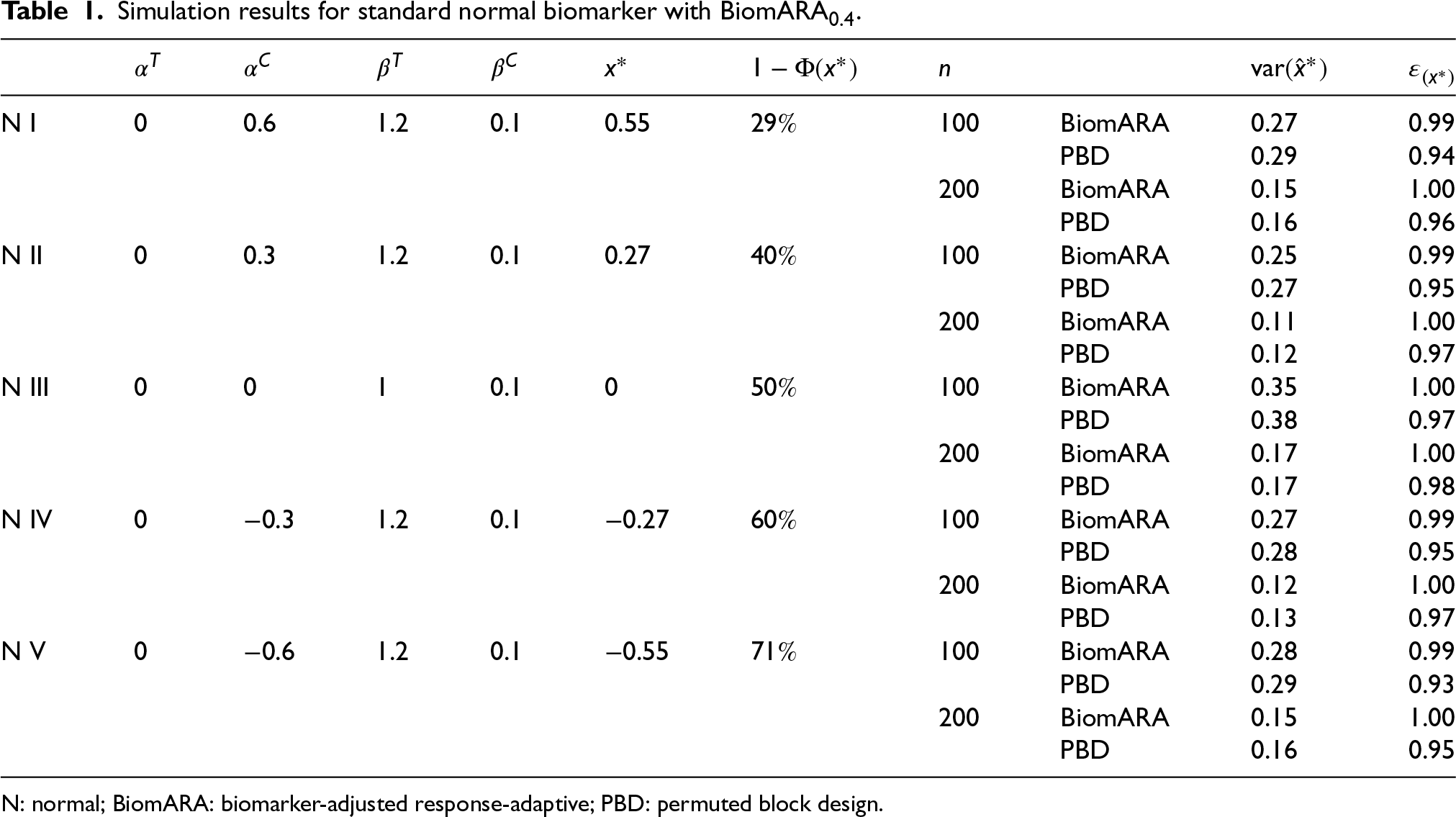
Focusing on inference on the threshold, we now consider five experimental scenarios (N I–N V), taking a standard normal biomarker with and . For each scenario (represented graphically in the left panel of Figure 4), in Table 1, we report , the true threshold and , namely the probability for a subject to have biomarker value greater than and so to benefit from (since ). From N I to N V, the value of decreases and thus the reported results refer to an increasing proportion of patients benefitting from , to mimic a range of possible real clinical settings. BiomARA and PBD are compared in terms of the empirical variance of the estimated threshold and the corresponding efficiency. In general, takes smaller values under our procedure with respect to those under PBD and BiomARA shows always higher values of that reach the maximum for .
Biomarker density function and success probabilities of (black curve) and (red curve) as varies. The displayed vertical lines correspond to the estimated median (grey dashed) and (green dashed).
Behavior of as increases for scenarios N II and LN II. N: normal; LN: log-normal.
Simulation results for standard normal biomarker with BiomARA.
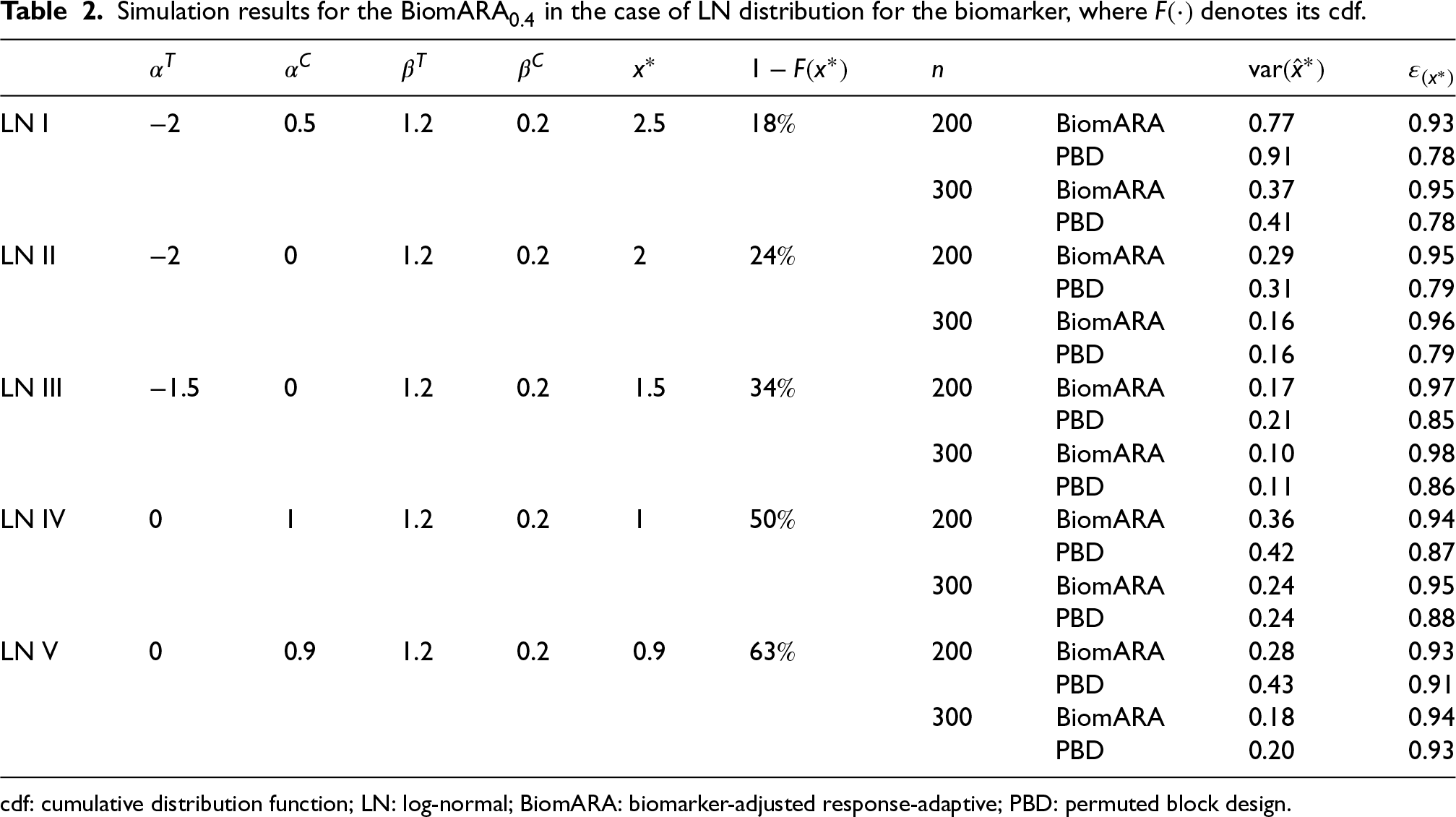
Under the LN distribution for the biomarker, we take into account five different scenarios (LN I–LN V), represented graphically in the right panel of Figure 4: the results are reported in Table 2 for and . With regard to , similar considerations to the normal distribution case hold, while seems to grow slower with , as already observed in the right panel of Figure 2.
Simulation results for the BiomARA in the case of LN distribution for the biomarker, where denotes its cdf.
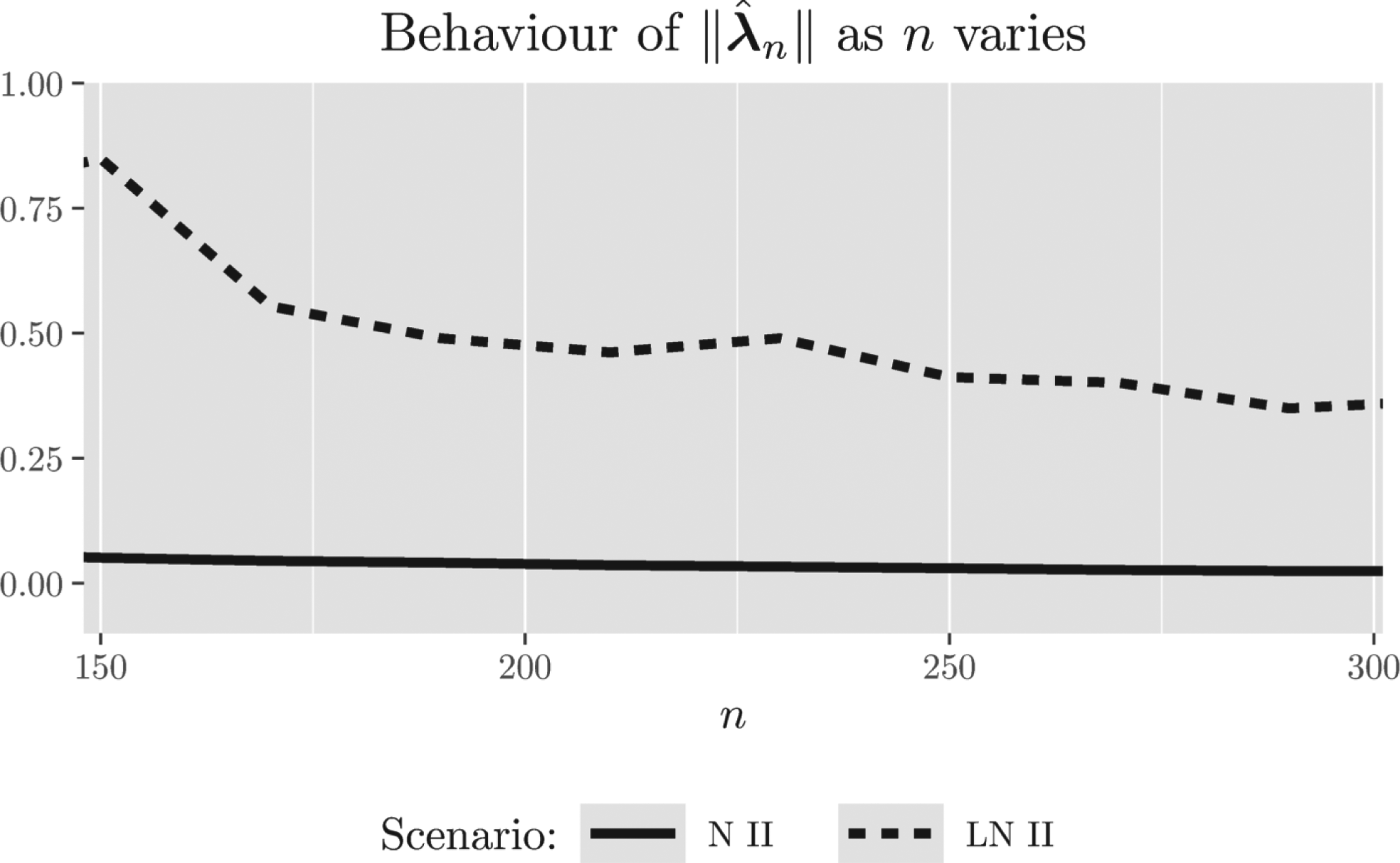
In general, the introduced BiomARA procedure converges to the suggested optimal designs, due to the independent and identically distributed nature of the covariate process that guarantees a stabilizing behavior for large samples (by the strong law of large numbers); however, the speed of convergence seems to be strongly related to the symmetry of the covariate distribution: Figure 5 shows the decreasing behavior of as grows for N II and LN II. The same graphical representation has been observed in all the other settings, omitted here for brevity, especially due to the slower convergence of the estimators in the asymmetric scenarios.
Note that, due to the variability in the estimation of (especially for small sample sizes or when is closer to the extremes of the support of the biomarker distribution), some of the simulated trials may end with the erroneous conclusion that no threshold exists (namely the estimated threshold is outside the range of variation of the biomarker). In these cases, the estimated proportion of patients with biomarkers greater than the threshold is one (the whole population benefits from ) or zero (nobody benefits from ). The results of Tables 1 and 2 are achieved by omitting those runs. In practice, the proportion of simulated trials leading to an erroneous conclusion depends on the number of subjects enrolled: clearly, the more patients are enrolled, the closer the estimated threshold will be to the true value, leading to a more accurate identification of the patient benefitting subpopulation. Indeed, preliminary clinical studies for identifying subpopulations require larger sample sizes than traditional nonpersonalized medicine trials (Mackey and Bengtsson21) and a metric based on this proportion has been used as a criterion to select the sample size (see Frieri et al.12). In the presented simulation study, we investigated only settings in which the percentage of reaching a wrong conclusion was lower than . Based on this, higher sample sizes have been set for the LN distribution.
The ethical impact of discretizing a continuous biomarker
In the presence of a continuous predictive biomarker, common practice has been to take its median to discriminate among the two subpopulations of biomarker-positive (sensitive) and biomarker-negative (insensitive) patients. Nevertheless, a clinical study to identify a threshold could be a preliminary stage for later phases and should determine some guidance for future use of and .6 In practice, if the true threshold would be a priori known, we would assign the treatment only to patients belonging to the sensitive subpopulation. We perform a simulation study to emulate such a situation in order to show the ethical impact of taking the data-driven median as a threshold instead of the one suggested in this paper, calculated via the relative risk.
To this end, we take into account the results obtained in Tables 1 and 2 (for ) as if they represent a preliminary phase through which the researcher had the opportunity to estimate both the median and the cutoff. Figure 4 describes the experimental scenarios N I–N V and LN I–LN V: the black (red) curve corresponds to the success probability of ()—namely ()—while the gray curve represents the standard normal density (left panel) and the LN density (right panel). Finally, the gray dashed line is the corresponding estimated median, while the green dashed line is (calculated by using the estimates of model parameters found in the first phase). We simulate patients’ biomarkers and we assign them to or by using (i) the estimated median and (ii) . Then, we compute the percentage of patients receiving the inferior treatment ( IT) when using the estimated median to discriminate the subpopulations, instead of . The results are reported in Table 3. Clearly, as also suggested by Figure 4, this proportion is very small for N III and LN IV–V, since is closer to the median. However, this percentage could grow up to for N and for LN, stressing that discretizing a continuous biomarker could lead to very serious ethical consequences and should be avoided.
Proportion of patients assigned to the inferior treatment if the estimated median is used instead of the estimated threshold based on the relative risk.
IT
N I
N II
N III
N IV
N V
IT
LN I
LN II
LN III
LN IV
LN V
% IT: percentage of patients receiving the inferior treatment; N: normal; LN: log-normal.
Redesign a clinical trial for severe sepsis disease
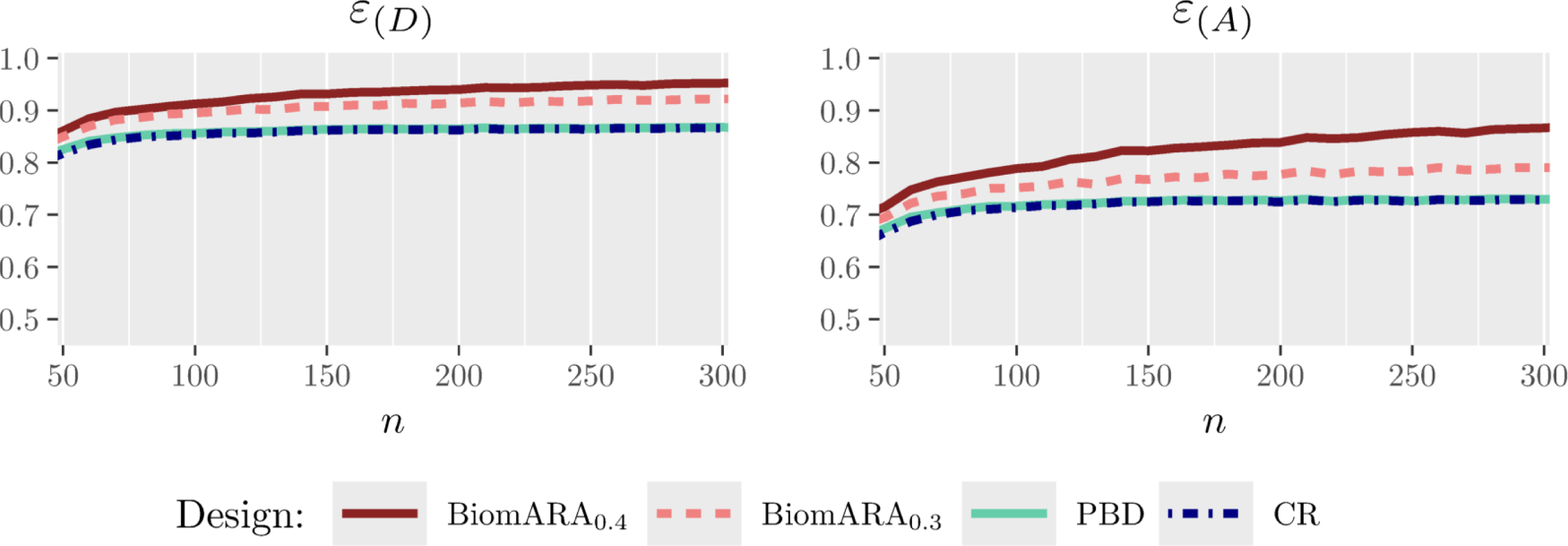
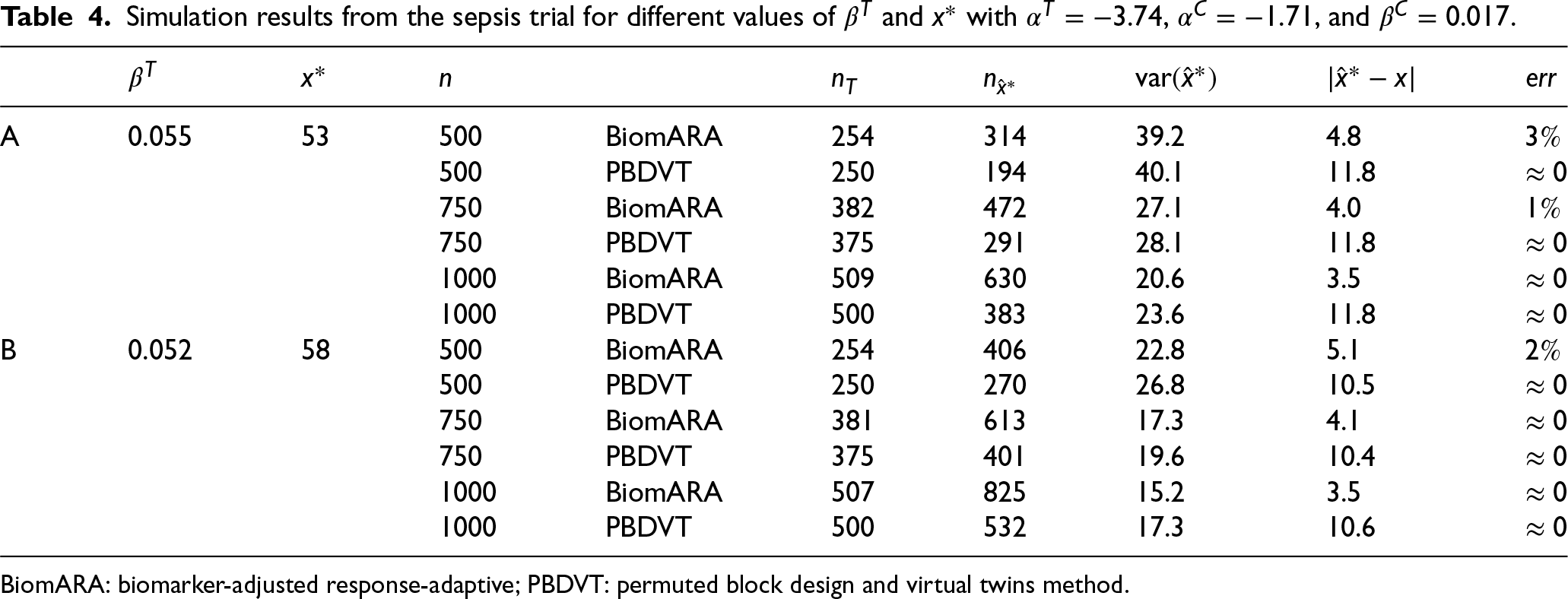
The following simulation study is intended to mimic a clinical trial for patients with severe sepsis disease. The primary outcome is status (deceased or not) at 28 days after treatment. Based on this dataset, since age is found to be a predictive biomarker, the goal is to find the age above which the success probability of the treatment is higher than the success probability of the control. The values of the age of patients, ranging from 33 to 93 years, were sampled with replacement from the dataset available at https://biopharmnet.com/subgroup-analysis-software/ while patients’ responses were simulated across different experimental scenarios corresponding to different underlying truths about the predictive strength of the biomarker and, consequently, the threshold. Scenario A refers to , , , and , obtained by fitting a logistic model to the data: in this case, the true threshold is around 53 years. Scenario B takes into account (the corresponding threshold is 58 years). In this study, the BiomARA (with ) is compared to a design strategy (Permuted Block Design and Virtual Twins method) which employs the PBD of size 4 for randomizing subjects and the Virtual Twins method1 as a subgroup identification method (implemented by the R package aVirtualTwins available on CRAN). For each scenario, Table 4 reports the average number of patients in the treatment group , the average number of patients with age greater than the estimated threshold , the average as a measure of bias and the percentage of simulations in which the trial ends up with a wrong conclusion. BiomARA shows smaller values of both the variance and the bias of the threshold, while the Virtual Twins method seems to suffer from a severe bias in this context. Finally, as previously discussed, is strongly sensitive to the sample size and to the closeness of the estimated threshold to the extremes of the support of the biomarker; however, due to the strong consistency of the MLEs, vanishes as increases regardless of the true threshold.
Simulation results from the sepsis trial for different values of and with , , and .
In this paper, we address the complex issue of patient enrollment restriction in the presence of a continuous predictive biomarker. Despite the advances in understanding disease mechanisms and the increasing discovery of biomarkers that affect patients’ responses to treatments, methodological results regarding continuous biomarker cutpoint identification and evaluation remain relatively few.
For binary response trials, in this work, we show that optimal designs for the estimation of the model parameters and the cutoff of a continuous predictive biomarker require multiple conditions to be satisfied, involving the patients’ biomarker values and the unknown model parameters. Such an optimal design can be implemented sequentially by adopting the new CARA procedure suggested in Section 4. An extensive simulation study, including a redesign of a clinical trial, highlights the advantages of the proposed approach.
Our research includes an analysis aimed at pointing out the pitfalls of the common and problematic approach of adopting (empirical) median-based biomarker cutoffs. From an ethical perspective, a fundamental requirement of a clinical trial is to ensure an overall benefit for the entire sample of enrolled patients and a widely used ethical criterion focuses on maximizing the percentage of patients who receive the best treatment. Given a prognostic biomarker (i.e. in the absence of treatment/covariate interactions), the relative performance of the treatments is the same for every subject’s profile; whereas, for predictive biomarkers (namely in the presence of treatment/covariate interactions), the superiority/inferiority of a given treatment, as well as their discrepancy, depends on the subject’s profiles. In this setting, discretizing continuous biomarkers is widely discouraged due to inferential and ethical repercussions (Polley and Dignam,3 Zhang and Molinaro,4 Bennette and Vickers,22 and Royston et al.23). In contrast, our approach provides a cutoff identification methodology directly on a continuous scale which guarantees that more patients are assigned to the superior treatment (based on their biomarker), in line with the International Code of Medical Ethics, “a physician shall act in the patient’s best interest when providing medical care.” We strongly believe that the discretization of continuous biomarkers should be avoided and we encourage the researchers to abandon it.
The suggested procedure is suitable for early phase studies as it is aimed at guiding future treatment decisions for individual patients. It is worth mentioning that the approach proposed in this paper can be applied to any continuous biomarker distribution, provided that widely satisfied assumptions hold.
Some potential directions for future research have been identified. For instance, extending our approach by considering the generalized linear model family would certainly expand its applicability to a broader spectrum of clinical trial scenarios. In addition, we intend to extend our procedure to accommodate more than two treatments. Lastly, yet importantly, incorporating multiple biomarkers could make the identification of the target population more accurate.
Footnotes
Acknowledgments
The authors of this paper wish to thank the Editor and the referees, who made substantial comments that improved the paper.
Declaration of conflicting interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: A. Baldi Antognini, R. Frieri and M. Zagoraiou were supported by the project Funded by the European Union—NextGenerationEU through the Italian Ministry of University and Research under the National Recovery and Resilience Plan (PNRR)—Mission 4 Education and research—Component 2 From research to business—Investment 1.1 Notice Prin 2022—DD N. 104 del 2/2/2022, title [Optimal and adaptive designs for modern medical experimentation], proposal code [2022TRB44L]—CUP [J53D23003270006]. A. Baldi Antognini and S. Cecconi were supported by European Union funding within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project No. PE00000007, INF-ACT).
ORCID iDs
Alessandro Baldi Antognini
Sara Cecconi
Rosamarie Frieri
Maroussa Zagoraiou
A. Appendix
B. Efficiencies
Now we derive the relative efficiency of a generic design compared to the optimal one in (6). Since the efficiency is
Since the efficiency is
As regards the threshold estimation,
so that the corresponding efficiency is
References
1.
FosterJTaylorJRubergS. Subgroup identification from randomized clinical trial data. Stat Med2011; 30: 2867–2880.
2.
LohWCaoLZhouP. Subgroup identification for precision medicine: A comparative review of 13 methods. Wiley Interdiscip Rev: WIREs Data Min Knowl Discov2019; 9: e1326.
3.
PolleyMYCDignamJJ. Statistical considerations in the evaluation of continuous biomarkers. J Nucl Med2021; 62: 605–611.
4.
ZhangYMolinaroAM. Categorizing continuous biomarkers: More cons than pros. Neurooncol Pract2022; 9: 81–82.
5.
JiangWFreidlinBSimonR. Biomarker adaptive threshold design: a procedure for evaluating treatment with possible biomarker-defined subset effect. J Natl Cancer Inst2007; 99: 1036–1043.
6.
Baldi AntogniniAFrieriRZagoraiouM. New insights into adaptive enrichment designs. Stat Pap2023; 64: 1305–1328.
7.
SpencerAHarbronCManderA, et al.An adaptive design for updating the threshold value of a continuous biomarker. Stat Med2016; 35: 4909–4923.
8.
TrippaLLeeEQWenPY, et al.Bayesian adaptive randomized trial design for patients with recurrent glioblastoma. J Clin Oncol2012; 30: 3258.
9.
RenfroLACoughlinCMGrotheyAM, et al.Adaptive randomized phase II design for biomarker threshold selection and independent evaluation. Chin Clin Oncol2014; 3: 3489.
10.
DiaoGDongJZengD, et al.Biomarker threshold adaptive designs for survival endpoints. J Biopharm Stat2018; 28: 1038–1054.
11.
LinZFlournoyNRosenbergerW. Inference for a two-stage enrichment design. Ann Stat2021; 49: 2697–2720.
12.
FrieriRRosenbergerWFlournoyN, et al.Design considerations for two-stage enrichment trial. Biometrics2022; 79: 2565–2576.
13.
Baldi AntogniniAFrieri RWRF, et al.Optimal design for inference on the threshold of a biomarker. Stat Methods Med Res2024; 33: 321–343.
14.
MargoleseRCecchiniRJulianT, et al.Anastrozole versus tamoxifen in postmenopausal women with ductal carcinoma in situ undergoing lumpectomy plus radiotherapy (NSABP B-35): a randomised, double-blind, phase 3 clinical trial. Lancet2016; 387: 849–856.
15.
VinnatVChevretS. Enrichment bayesian design for randomized clinical trials using categorical biomarkers and a binary outcome. BMC Med Res Methodol2022; 22: 54.
16.
SpreaficoAHansenAAbdul RazakA, et al.The future of clinical trial design in oncology. Cancer Discov2021; 11: 822–837.
17.
AzoulayELemialeVMokartD, et al.Effect of high-flow nasal oxygen vs standard oxygen on 28-day mortality in immunocompromised patients with acute respiratory failure: The HIGH randomized clinical trial. JAMA2018; 320: 2099–2107.
AtkinsonACDonevANTobiasR. Optimum experimental designs, with SAS. Oxford: Oxford University Press, 2007.
20.
MaZHuF. Balancing continuous covariates based on kernel densities. Contemp Clin Trials2013; 34: 262–269.
21.
MackeyHBengtssonT. Sample size and threshold estimation for clinical trials with predictive biomarkers. Contemp Clin Trials2013; 36: 664–672.
22.
BennetteCVickersA. Against quantiles: categorization of continuous variables in epidemiologic research, and its discontents. BMC Med Res Methodol2012; 12: 1–5.
23.
RoystonPAltmanDSauerbreiW. Dichotomizing continuous predictors in multiple regression: a bad idea. Stat Med2006; 25: 127–141.