
Abstract
In the search for effective treatments for COVID-19, the initial emphasis has been on re-purposed treatments. To maximize the chances of finding successful treatments, novel treatments that have been developed for this disease in particular, are needed. In this article, we describe and evaluate the statistical design of the AGILE platform, an adaptive randomized seamless Phase I/II trial platform that seeks to quickly establish a safe range of doses and investigates treatments for potential efficacy. The bespoke Bayesian design (i) utilizes randomization during dose-finding, (ii) shares control arm information across the platform, and (iii) uses a time-to-event endpoint with a formal testing structure and error control for evaluation of potential efficacy. Both single-agent and combination treatments are considered. We find that the design can identify potential treatments that are safe and efficacious reliably with small to moderate sample sizes.
Introduction
The emergence of COVID-19 and the ensuing pandemic has led to a widespread, frantic, search for treatments. Despite large uncertainty about the underlying pathogen and the natural history of the disease, trials must start rapidly to identify treatments to save lives, but also so that effective treatments can be used in the response to the outbreak. A consequence of this is that trials in COVID-19 in the first few weeks and months of the outbreak have focused on re-purposed treatments.1–3
While recently, some success with using re-purposed treatments has been achieved,4–6 it is crucial that development of treatments specifically developed for COVID-19 is also undertaken in order to maximize the chances of finding therapies to successfully treat patients. The crucial difference of trials investigating novel therapies (in contrast to re-purposed treatments) is that the range of safe and likely effective doses is unknown. Therefore, an efficient dose-finding design identifying safe and active doses to be studied in larger trials is essential. While there exist a number of dose-finding designs for early phase dose-finding trials evaluating toxicity and efficacy simultaneously, for example, Wages and Tait 7 and Mozgunov and Jaki 8 and references therein, many of them consider a binary efficacy endpoint with few recent extension to other endpoints.9–11 Time-to-event endpoints with censoring at 28 days have previously been used as a clinically meaningful measure in a number of COVID-19 trials1,2,4 and the argument has been made that they should be considered in all COVID-19 trials. 12
While the majority of Phase I dose-finding trials, particularly in oncology, are non-randomized, it is agreed that in later phases, the gold standard for evaluating novel treatments are well-conducted blinded randomized controlled clinical trials. At the same time, in light of the uncertainty about the symptoms caused by COVID-19 – especially at the beginning of the pandemic – it is essential to conduct randomized dose-finding trials to ensure that the risk of adverse events is correctly attributed to the drug under study rather than to the disease itself. Moreover, it has been argued that adaptive designs13,14 are particularly suitable during a pandemic, also in the light of the uncertainty about a novel disease. 15 Therefore, a randomized adaptive dose-finding design evaluating both toxicity and time-to-event efficacy would allow to answer the research questions of interest in novel therapies for treating COVID-19.
It is also recognized that there are a number of novel therapies that have the potential to be efficient in fighting COVID-19. Therefore, it is crucial to have a structure in place that allows rapid enrolment of novel therapies to ensure rapid decision-making, and, importantly, would allow for efficient use of information between the studies, that is, utilizing the data from the control treatment across different compounds. This can be achieved via a platform trial. 16
In this paper, we describe and evaluate the bespoke design developed and implemented for the AGILE platform,
17
an adaptive randomized seamless Phase I/II dose-finding trial platform that seeks to quickly establish a safe range of doses and investigates treatments for potential efficacy using a Bayesian sequential trial design (see a visualisation of the design for one compound in Figure 1). The proposed design is unique as it
utilizes randomization during dose-finding to allow COVID-19 induced symptoms to be distinguished from drug side-effects, shares control arm information across the platform in order to maximize efficiency, and uses a time-to-event endpoint with a formal testing structure and error control for evaluation of potential efficacy,
making the design particularly suitable for the pandemic setting. We also extend the design for trials studying dual-agent combinations of treatments.
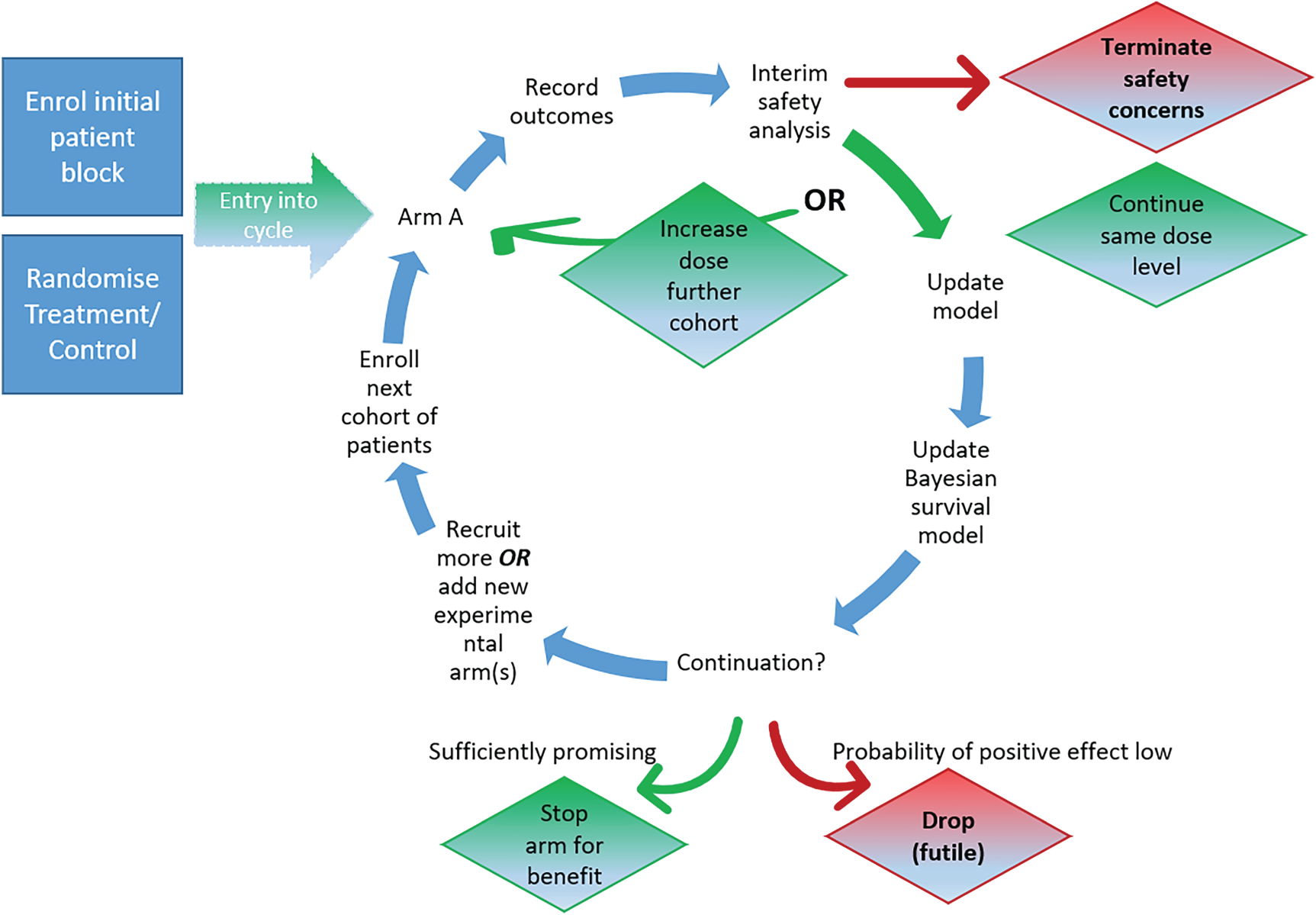
Illustration of the AGILE platform design.
The rest of the article is organized as follows. Section 2 describes the platform for single treatments while its performance is evaluated in simulations in Section 3. The design for dual-agent combinations is proposed in Section 4 and subsequently evaluated in Section 5. We conclude with a discussion (Section 6).
Setting
Consider a randomized controlled dose-escalation clinical trial in which
As it is expected that the control arm is associated with a non-negative (unknown) risk of DLE (or symptoms of the disease that cannot be distinguished from DLEs), the primary goal of the dose-escalation is formulated in terms of the additional risk of a dose-limiting event (ADLE) defined in terms of the expected difference in DLE risk between the doses of the agent and the control. Specifically, we therefore seek to identify the dose that corresponds to an additional risk of
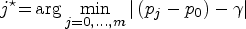
Bayesian dose-escalation model
The following randomized Bayesian dose-escalation design that builds on the proposal by Mozgunov et al.
18
is used. Assume that the DLE probability has the functional form
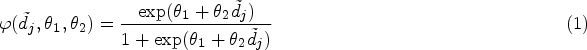
Denote the prior distribution of the vector

Assume that
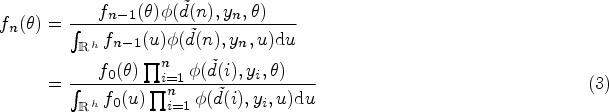


Bayesian efficacy model
In this study, we assess the potential efficacy of the treatment for a particular dose instead of modelling efficacy across all doses. Although other approaches are possible, our approach allows us to make conclusions about a given dose alone without sharing information from other arms and enables control of the type I error for the assessment of a given dose. A Cox proportional hazards model is assumed where the hazard of recovery at time
A Bayesian criterion is adopted for the stopping rule at each stage. In line with Bayesian thinking, we set the stopping rules to be the same for each stage evaluation is stopped for efficacy if evaluation is stopped for futility if an additional cohort of patients is recruited, otherwise.
In order to ensure a decision is made at the final stage
A point prior of the form
An advantage of the point prior is that obtaining the posterior probability
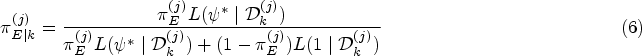
To set the boundaries,
The effect of varying
The inclusion of historic controls will increase both the power and type I error of any procedure, for example, Schmidli et al.
22
To ensure type I error is controlled for the evaluation of the given dose, the boundaries are set assuming the maximum,
Overall design
The overall design of the platform allows for multiple different compounds to be evaluated and, by sharing concurrent control group data, efficiency is gained. For any compound in the platform, patients are allocated in cohorts of size
Safety evaluation
The first cohort of After The set of safe doses is found using equation (4).
If no doses are safe, the trial is stopped for safety; if only the current dose is safe, the next cohort of otherwise, the next cohort of Once efficacy information is available for two cohorts on a safe dose, that dose is graduated to the efficacy evaluation. If a dose The posterior probability, If if otherwise if
Efficacy evaluation
The evaluation of a dose continues until the maximum number of patients
Evaluations of proposed design
Setting
We will now evaluate, for one compound, safety and efficacy across the study together in a simulation study and evaluate the impact of shared control data that are gradually accumulated over the course of the trial, thereby assessing the added benefit of the platform structure. To our knowledge there are no alternative approaches that (i) utilize randomization during dose-finding, (ii) assess efficacy using a time-to-event endpoint within a formal testing framework and (iii) employ a platform structure and hence no comparator is presented here. Our sensitivity analysis presented in the supplemental materials, however, provides a comparison with a design that uses a similar design with a binary outcome for efficacy.
We consider the setting where there are three active doses (
The maximum total intake per dose level is 72 patients assigned to each dose level and control, which equates to a maximum total sample size of 216. In line with the real study, cohort sizes of
The objective of the trial is to find all safe efficacious doses to be graduated into a larger Phase II or Phase III clinical trial. The target ADLE risk is
Scenarios
As the trial aims to study novel compounds which have yet to be explored with respect to their mechanism of action in COVID-19 patients, it is crucial that the design has good operating characteristics under a variety of dose-DLE and dose-efficacy scenarios. Therefore, we consider five dose-efficacy scenarios ranging from no doses corresponding to a change in time-to-improvement within 28 days to all doses resulting in a clinically significant reduction; and five dose-DLE scenarios ranging from all doses being safe to all doses being very unsafe. We then consider all combinations of these scenarios, resulting in 25 scenarios explored in total. The five dose-DLE and dose-efficacy scenarios for each (
Safety and efficacy scenarios for (
,
,
, and
).
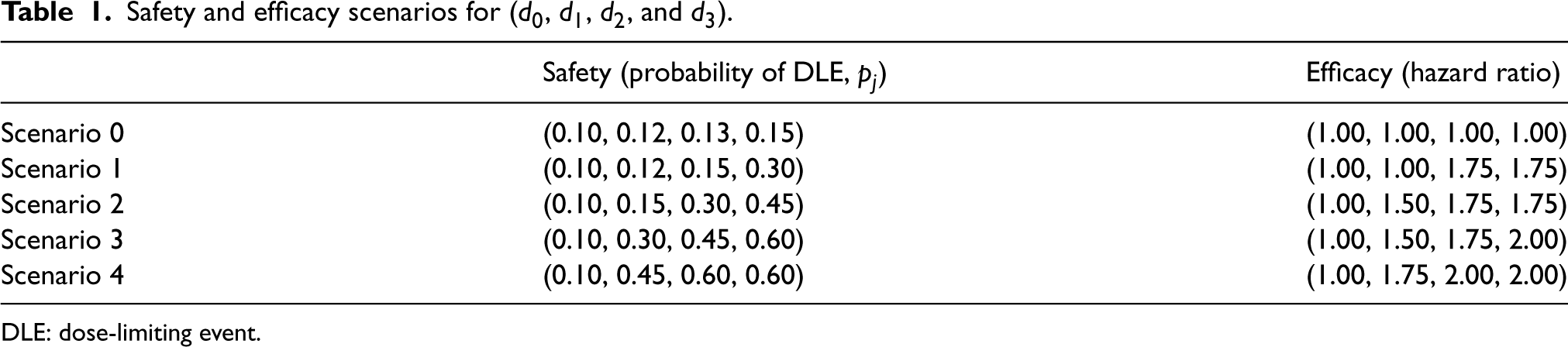
Safety and efficacy scenarios for (
DLE: dose-limiting event.
We will refer to the scenario with dose–DLE relationship
For all 25 scenarios, a sensitivity analysis is conducted on varying values of
Software in the form of R code used to produce the presented results is available on GitHub (https://github.com/dose-finding/covid19-agile).
Safety model
The proposed design requires the prior and design parameters for both safety and efficacy parts to be pre-specified in advance of the conduct of the trial. The procedure of how these parameters were chosen is given below.
The prior parameters for the safety model were obtained via a calibration procedure
25
over a number of safety scenarios (not taking into account efficacy). We use safety Scenarios 1 to 3 in Table 1 that correspond to the target dose being
The following prior distribution for the vector of safety model parameters 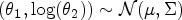
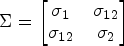
Furthermore, to define the standardized doses
The calibration was performed as follows. For each combination of parameters of
Efficacy model
The efficacy stopping boundaries for a particular setting were taken as the pair 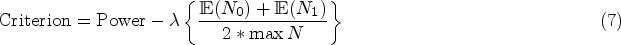
A value of
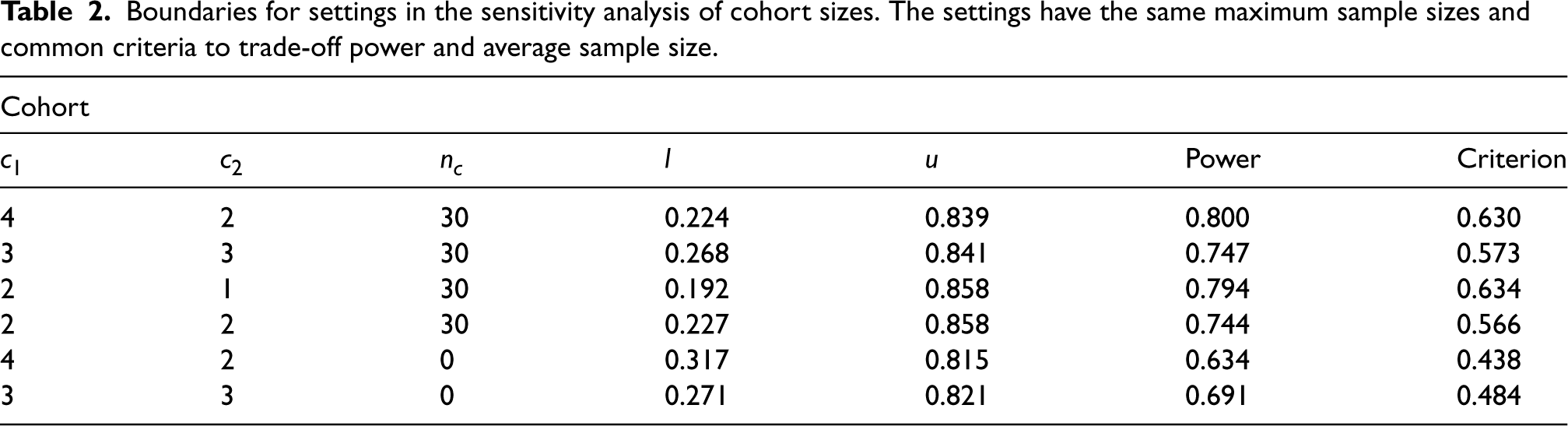
Boundaries for settings in the sensitivity analysis of cohort sizes. The settings have the same maximum sample sizes and common criteria to trade-off power and average sample size.
Throughout, the point prior is taken to be 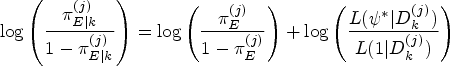
Detailed results for the setting with a cohort size of
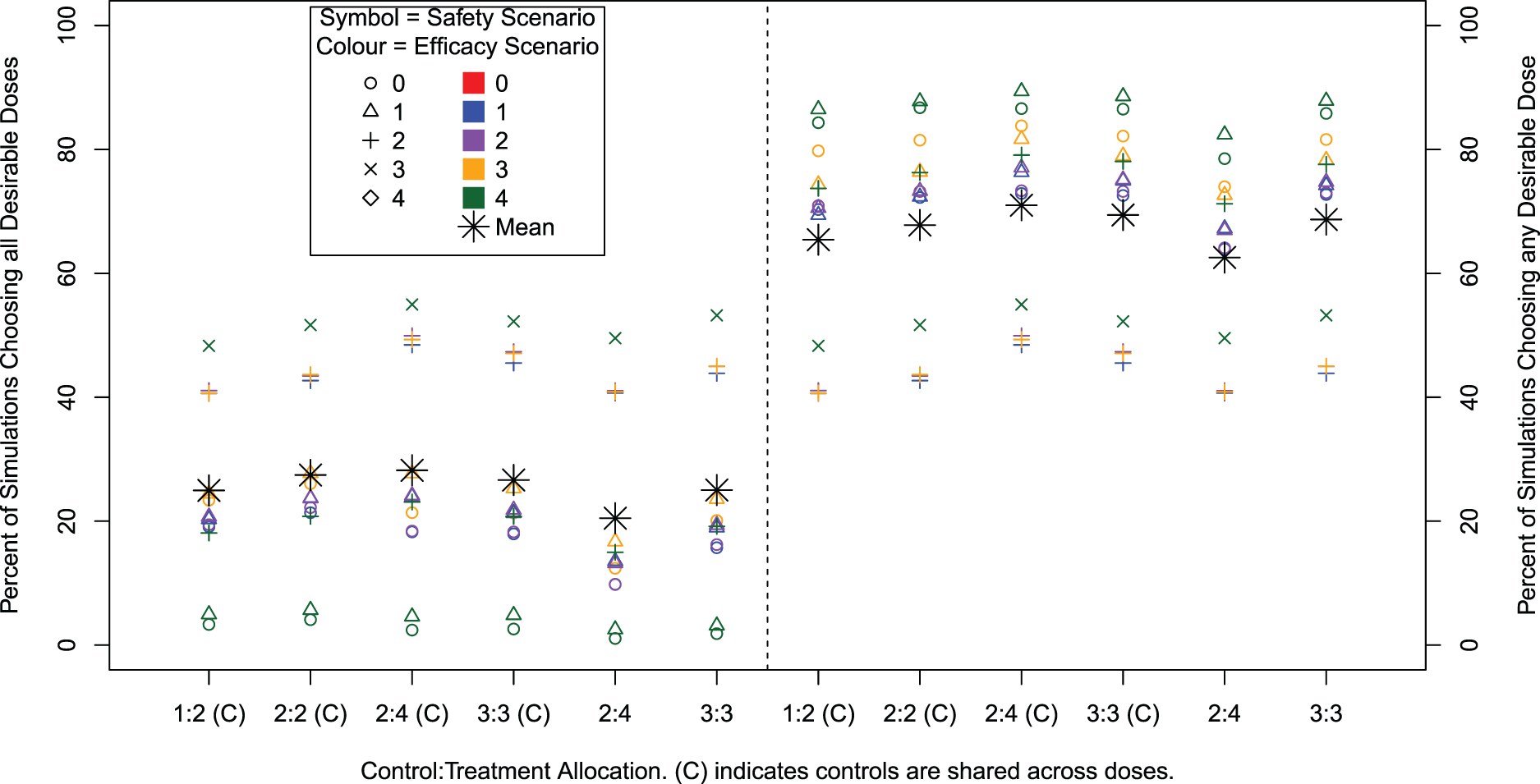
Percentage of simulations that recommend all desirable doses (left) and the percentage of simulations that recommend any desirable dose (right) for different cohort sizes and compositions and with and without sharing control group data. Note that only 13 out of 25 efficacy/safety scenarios contain a desirable dose.
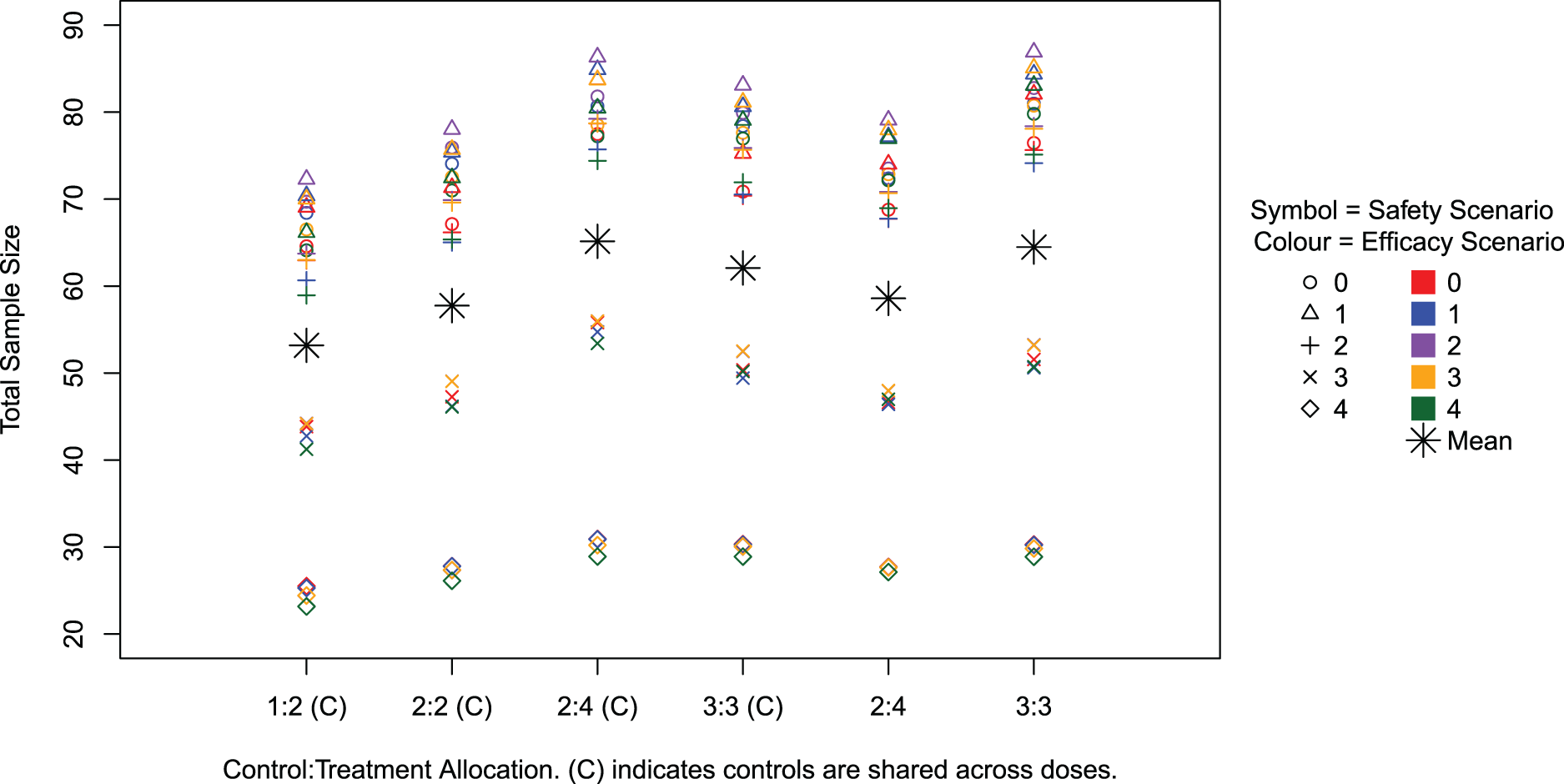
Average total sample size across simulations for all scenarios.
Percentage of 10,000 simulations where each dose is recommended for (
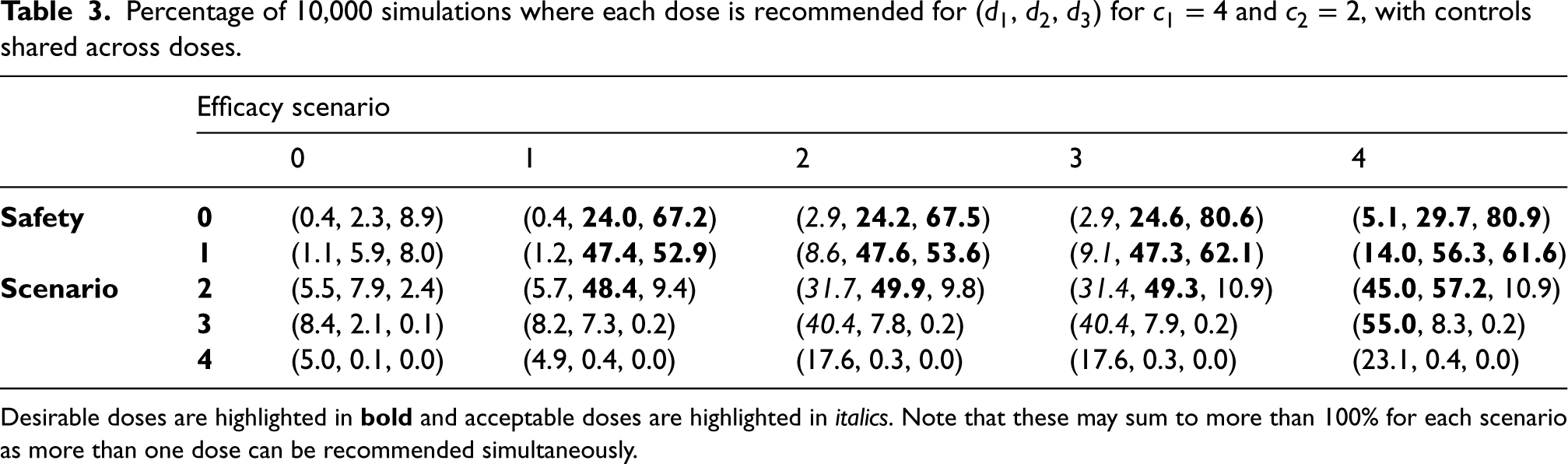
Desirable doses are highlighted in
Figure 2 shows the percentage of simulations where all desirable doses are recommended (left) and where any desirable doses are recommended (right). For the baseline setting of
The sensitivity analysis across the varying cohort settings shows there is a very small difference in performance. The ordering of performance across safety/efficacy scenarios is identical with only a small numerical difference. However, we can see that not sharing controls decreases the performance. Power for recommending any desirable dose increases for increasing cohort size
Figure 3 illustrates the average total sample size across the scenarios and settings. On average about 65 patients are required in the setting used in the trial with the total sample size exceeding 150 in only 1% of simulations across scenarios. The scenarios with the smallest sample sizes are those where all doses are unsafe and the trial is therefore stopped early for safety. In such cases, it takes 30 patients across 6 weeks on average to reach the conclusion of stopping early for safety. The scenarios with larger sample sizes are those where all doses are safe and most are acceptable or only just desirable (i.e. not the case where the hazard ratio is 2.00), as in these cases more doses are taken to the efficacy part and more patients are required to detect the smaller difference in hazard ratios.
It can be seen across settings that the larger the total cohort size, the larger the total sample size. This also corresponds to the higher power settings. When controls are shared, altering the control
Table 3 gives more detail into which doses are recommended across simulations. For example, in efficacy Scenario 2, where the lowest dose is acceptable and the higher two are desirable in terms of efficacy. In safety Scenario 0, where all doses are safe, the highest dose is chosen most often. In safety Scenario 2 where only the highest dose is unsafe, the middle dose is chosen most often, although less often than the highest desirable dose in Scenario 0. In safety Scenario 4 where all doses are unsafe, the lowest dose is chosen only 17.6% of the time. It is clear that desirable doses are recommended most often, with incorrect and undesirable doses rarely recommended. This gives insight that the procedure is successful in identifying desirable doses of a single agent.
Our additional sensitivity analyses investigating the violation of proportional hazards and providing a comparison against a binary efficacy outcome, presented in the supplemental materials, show that the design is fairly robust to violations of the proportional hazards assumption. Major violations of this assumption yield increased power at the expense of higher type I error. At the same time, these analyses show that the proposed time-to-event approach is superior to using a binary efficacy endpoint as expected.
Setting
Consider now a randomized controlled dose-escalation dual-agent clinical trial studying the combinations of 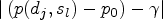
Dual-agent Bayesian dose-escalation model
For the considered randomized dual-agent combination setting, under the assumption of independence of the compounds, the probability of a DLE associated with combination 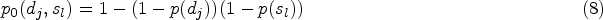
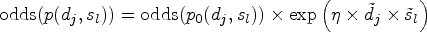
Note that 

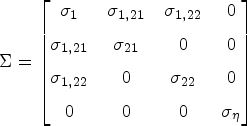
Parameters of the vector 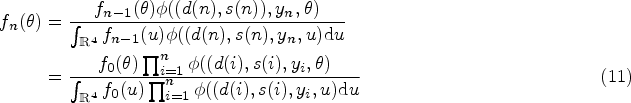
This posterior distribution is then used to make the escalation and de-escalation decision during the trials as proposed below.
The above combination-DLE model is then used in the design in Section 2.4 in place of the single-agent model. As in the single-agent setting, escalation can only occur to adjacent doses. As a consequence, no dose skipping is allowed and only escalation of one agent in the combination is permitted. In the case of equal probability for two eligible combinations, randomization is used. As the efficacy part of the dose-escalation design proposed for monotherapies considered each dose individually, the efficacy part of the combination study remains the same. Once the combination of the compounds is established to be safe, it is graduated into the efficacy part following the single-agent proposal and the same decision rules for dropping for futility and safety.
Evaluation of combination treatment design
Scenarios
In order to evaluate the dual-agent design, we conduct a simulation study comprising scenarios with two dose levels of agent
Safety and efficacy scenarios for dual-agent combinations
and
. It is assumed the control arm remains with a probability of dose-limiting event (DLE) 0.10 and a hazard ratio 1.00.
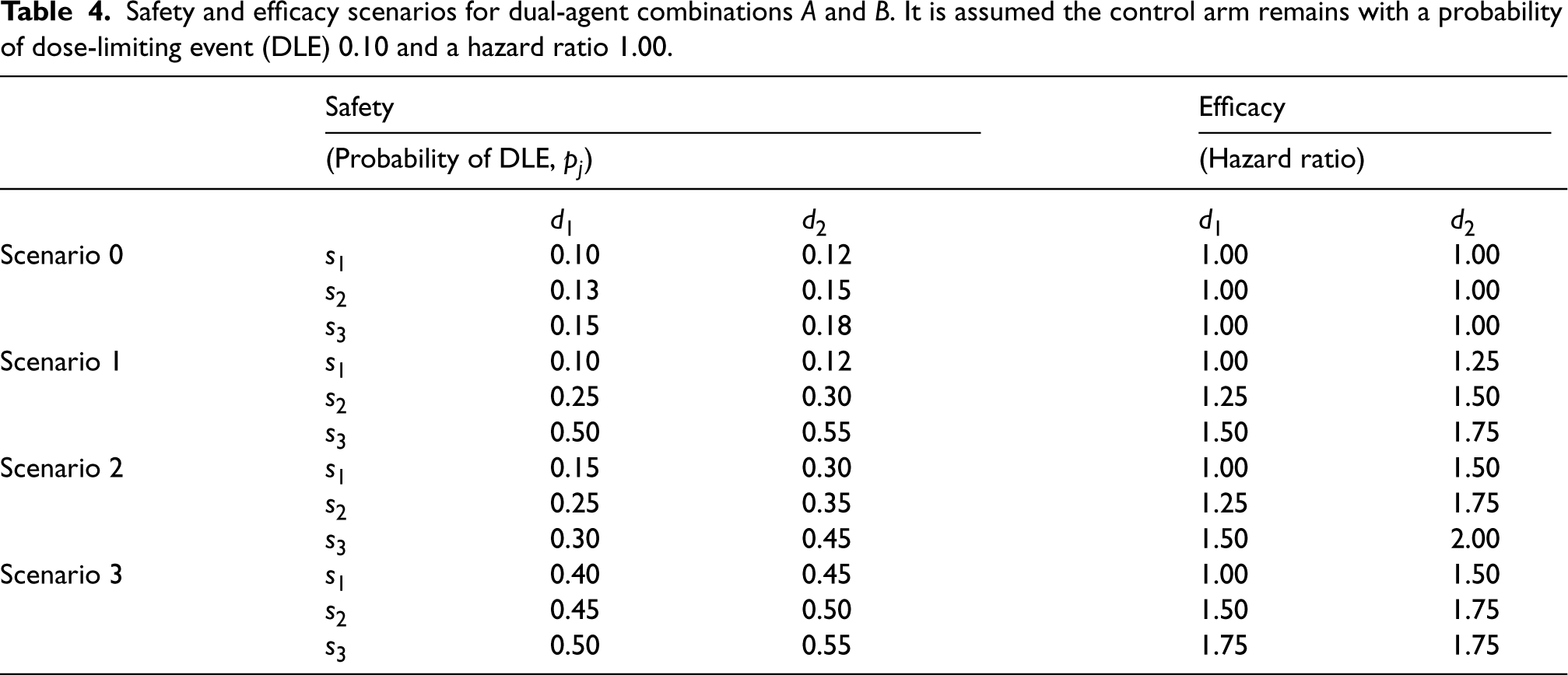
Safety and efficacy scenarios for dual-agent combinations
To define the parameters of the combination model, a calibration procedure similar to the procedure described in Section 3.3.1 was applied. Safety Scenarios 0 to 3 in Table 4 that correspond to different steepness of the combination–toxicity relationship and different locations of the target combination were used. We then choose the hyperparameters for the prior distribution of the parameters of the model,
Given the link between the prior toxicity on the control arm and the intercept parameter
As in the single-agent setting, the standardized doses
Using 500 simulations under each scenario and each combination of hyperparameters, the values
Results
For the 16 scenarios considered in the simulation study, the percentage of simulations recommending any desirable dose combination, the percentage of simulations recommending all correct dose combinations and the mean total sample size are presented in Figure 4, with further detail on individual dose combination recommendations given in Table 5. The overall type I error rate that is the percentage of simulations recommending any dose combination in Scenario 0–0, is 12.1%. By construction, the type I error for a given dose is controlled at 10%. In the extension from monotherapies to dual-agent therapies, some similar patterns are maintained in the results although there are some notable differences.
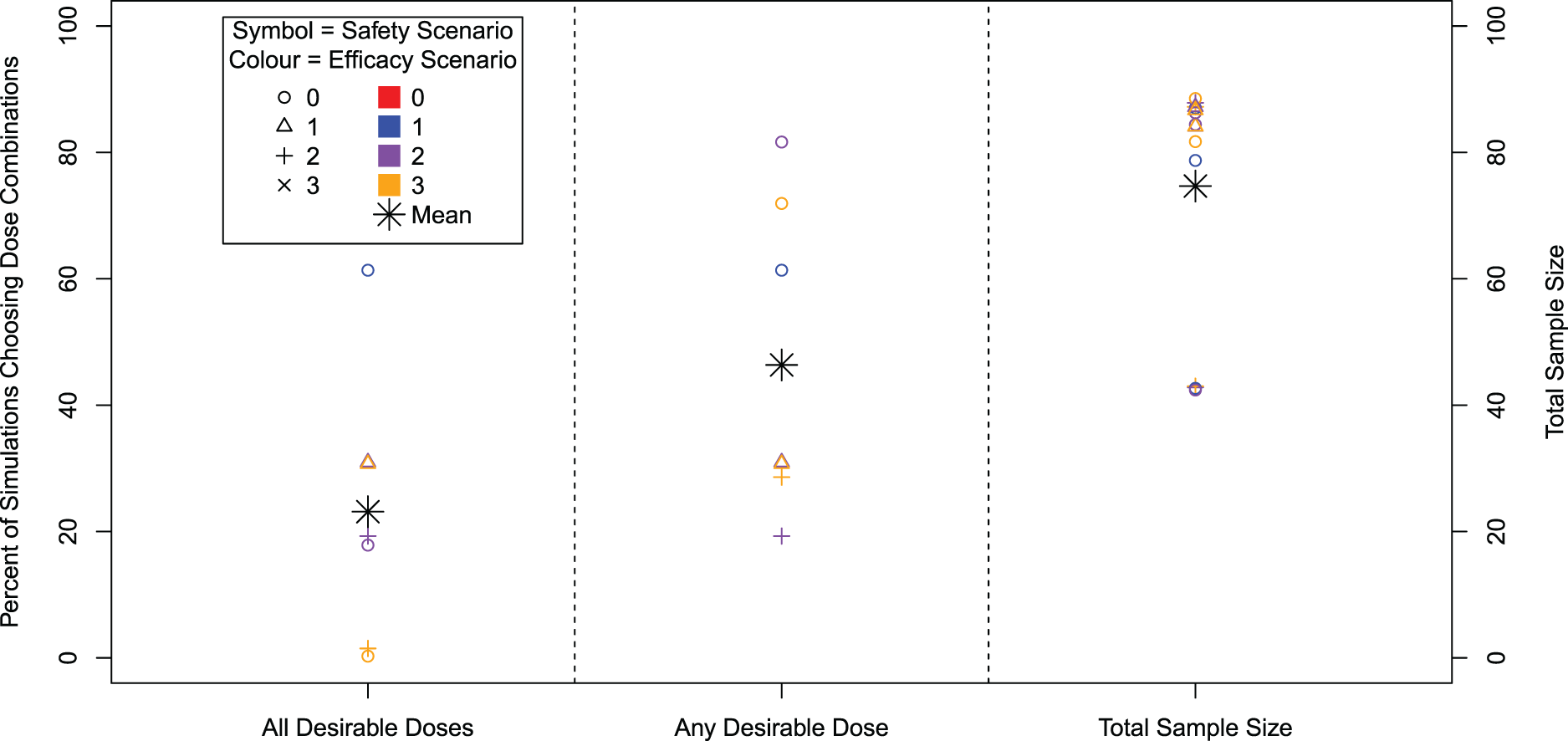
Percentage of 10,000 simulations that recommend all desirable dose combinations (left), the percentage of simulations that recommend any desirable dose combination (centre) and average total sample size. Note that only 7 out of 16 efficacy/safety scenarios contain a desirable dose combination.
Percentage of 10,000 simulations where each dose combination is recommended.
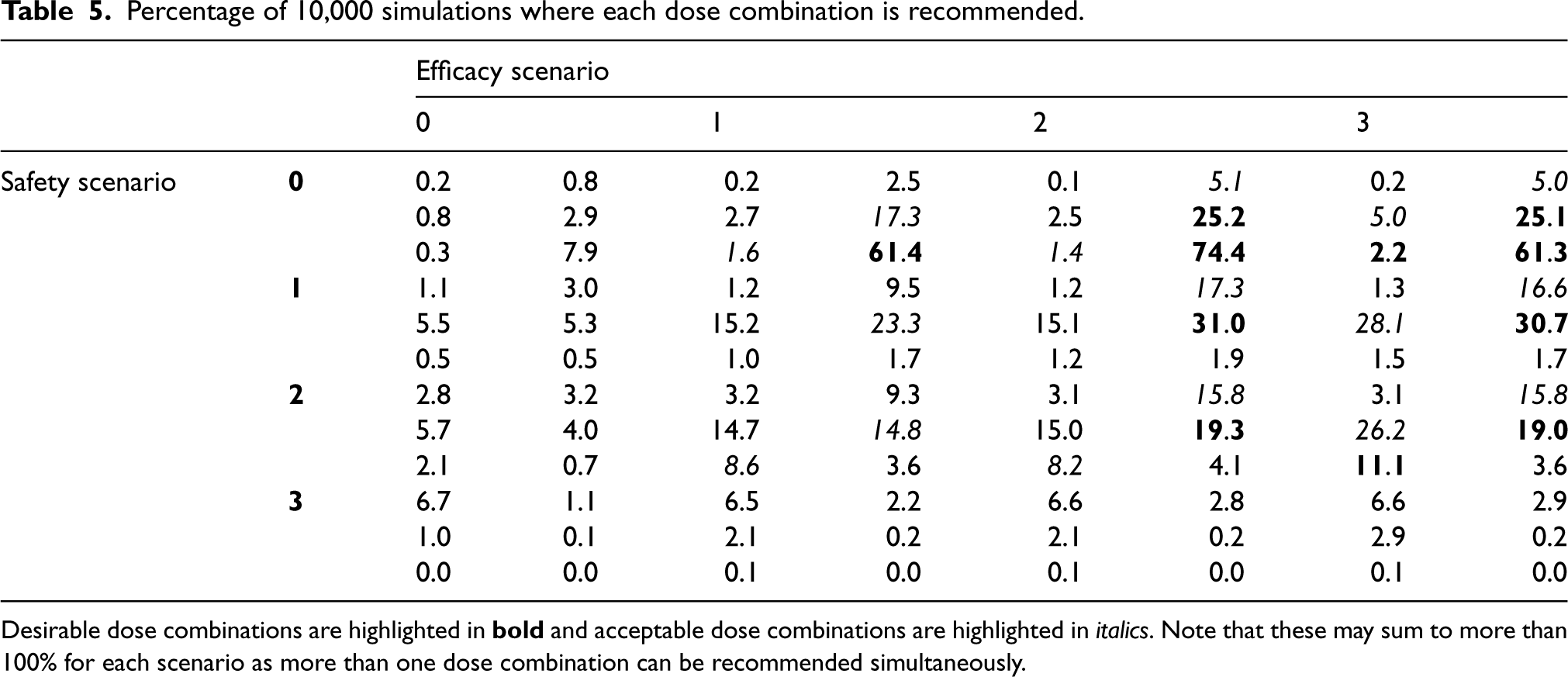
Desirable dose combinations are highlighted in
It can be seen in Figure 4 that the spread of powers across scenarios for dose combinations is larger than for monotherapies, for both the selection of all and any desirable dose combinations. The maximum power of 81.7% to select any desirable dose combination is achieved in Scenarios 0 to 2, where all doses are safe and there is a steep monotonic relationship within agents for efficacy. Even though there is an extra desirable dose combination in Scenarios 0 to 3, we observe a slightly reduced power since the most efficacious dose combination has a lower hazard ratio. The power to recommend all desirable doses ranges up to 61% in Scenario 0 to 1 where only one dose is desirable, with the lowest in Scenarios 0 to 3 and 2 and 3 where the desirable dose combinations are across separate agents’ dose-escalation (i.e. to select all desirable doses requires a de-escalation in one agent and then escalation in the other agent).
Across scenarios, the mean total sample size is 75, ranging from 42 to 89, a narrower range than for the single agent. However similar to the single agent, the smaller sample sizes correspond to scenarios where all dose combinations are unsafe and therefore the trial stopped early for safety. When this is not the case, there is little variation across scenarios in terms of mean total sample size.
Table 5 shows further details of dose recommendations in the simulations. Especially of note is the emphasis on recommendations of acceptable doses. For example, in Scenarios 2 and 3, where the power to detect desirable dose combinations is low, a large proportion of simulations also recommend an acceptable dose combination. It is also clear that inefficacious and/or unsafe doses are rarely recommended.
We introduce and evaluate the bespoke statistical design of the AGILE platform which seeks to quickly establish safe doses and potential for efficacy. The novel design utilizes a platform structure that allows the sharing of control data, includes a randomized dose-finding component and yields well-powered decisions about the activity of the treatments while controlling the type I error. We find that the design can identify potential treatments with good accuracy and show that the approach is easily extended to combinations of treatments.
The design uses a recently proposed randomized dose-finding design to ensure that differences between symptoms of COVID can be distinguished from side effects of the investigated treatment while a very simple Bayesian model is used to capture the potential efficacy of the treatments. The latter is in line with the objective of the trial: make reliable decisions about potential quickly, rather than using more complex methods that allow more precise estimation. At the same time, this approach guarantees that the whole platform structure can be simulated quickly to enable the study design to be fixed quickly.
The design has been constructed in a flexible manner using a time-to-event outcome and we based our simulations on time-to-improvement – an endpoint that has been shown recently to be a highly powered and relatively easy to collect. 12 The platform has, however, been constructed to also be able to investigate mild disease in which case a primary endpoint used would be time-to-negative viral titres in nose and/or throat swab. Provided that the event rate in this setting is the same, we expect that the performance reported here will be similar.
In line with Yeung et al., 28 we have opted for separate models for safety and efficacy to allow the timing of information assessment on safety (7 days) and efficacy (28 days) to be different in order to increase the speed of the dose-escalation. At the same time, Cunanan and Koopmeiners 29 found that in their evaluations using a joint model did not yield improved performance.
In setting up the AGILE platform and more generally when considering Phase I/II trials, several important choices, such as error rates, power and sample size need to be made. Given the exploratory nature of such studies, we believe that, in light of the small sample sizes, it is preferable to allow a somewhat larger type I error in order to achieve adequate power which will prevent missing potentially useful treatments at this early stage of development, something previously highlighted by Lindborg et al. 30 Future development will seek to extend the design to more general prior distributions for the efficacy model and consider extensions that allow the duration of treatment to be explored in addition to dose.
Supplemental Material
sj-pdf-1-smm-10.1177_09622802241288348 - Supplemental material for A seamless Phase I/II platform design with a time-to-event efficacy endpoint for potential COVID-19 therapies
Supplemental material, sj-pdf-1-smm-10.1177_09622802241288348 for A seamless Phase I/II platform design with a time-to-event efficacy endpoint for potential COVID-19 therapies by Thomas Jaki, Helen Barnett, Andrew Titman and Pavel Mozgunov in Statistical Methods in Medical Research
Footnotes
Acknowledgements
The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health and Social Care (DHSC). For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising.
Data availability
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). This report is independent research supported by the National Institute for Health Research (NIHR Advanced Fellowship, Dr Pavel Mozgunov, NIHR300576; and Professor Jaki’s Senior Research Fellowship, NIHR-SRF-2015-08-001). T Jaki, H Barnett and P Mozgunov also received funding from the UK Medical Research Council (MC_UU_00002/14, MC_UU_00040/03).
Supplemental material
Supplemental material for this article is available online.
A sensitivity analysis for the assumption of proportional hazards is available as part of the online article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.