
Abstract
In recent decades, many phase II clinical trials have used survival outcomes as the primary endpoints. If radiotherapy is involved, the competing risk issue often arises because the time to disease progression can be censored by the time to normal tissue complications, and vice versa. Besides, many existing research has examined that patients receiving the same radiotherapy dose may yield distinct responses due to their heterogeneous radiation susceptibility statuses. Therefore, the “one-size-fits-all” strategy often fails, and it is more relevant to evaluate the subgroup-specific treatment effect with the subgroup defined by the radiation susceptibility status. In this paper, we propose a Bayesian adaptive biomarker stratified phase II trial design evaluating the subgroup-specific treatment effects of radiotherapy. We use the cause-specific hazard approach to model the competing risk survival outcomes. We propose restricting the candidate radiation doses based on each patient’s radiation susceptibility status. Only the clinically feasible personalized dose will be considered, which enhances the benefit for the patients in the trial. In addition, we propose a stratified Bayesian adaptive randomization scheme such that more patients will be randomized to the dose reporting more favorable survival outcomes. Numerical studies and an illustrative trial example have shown that the proposed design performed well and outperformed the conventional design ignoring the competing risk issue.
Keywords
Introduction
A conventional phase II clinical trial tests whether the experimental drug has any anti-disease activity. The short-term efficacy outcome, such as the objective tumor response, is commonly used as the primary endpoint for a phase II clinical trial. Then, suppose the experimental drug shows sufficiently favorable short-term efficacy responses in a phase II trial. In that case, a large-scale phase III trial will be followed to test the long-term therapeutic effect using survival outcomes such as overall survival or progression-free survival (PFS). This widespread clinical practice assumes that the short-term efficacy outcome is an excellent surrogate marker for the long-term survival outcome. This assumption, however, does not always hold. For example, complete remission (CR) is the most desirable short-term efficacy outcome. However, achieving CR is necessary but insufficient for prolonging survival because many patients may relapse shortly after achieving CR. Indeed, many cytotoxic agents report favorable CR rates in phase II trials. However, only a few can transform improving CR rates into a substantial survival benefit in the following phase III trials. 1 Hence, to resolve this issue, in recent decades, there has been a growing trend to use the survival outcome as the primary endpoint for phase II clinical trials.2–6 This paper studies the phase II clinical trial design using survival outcomes, focusing on radiotherapies (RTs).
The RT is a “double-edged sword” for cancer patients. On the one hand, the X-ray on tumor cells can prevent disease progression; on the other hand, the X-ray on normal cells can induce normal tissue complications such as severe and irreversible organ damage (fibrosis, vascular damage, atrophy, etc.).7,8 Therefore, although the dose-limiting toxicity (DLT) has already been evaluated in phase I dose-finding trial, the normal tissue complications still need to be monitored in the phase II trial because (a) the normal tissue complications can be fatal, (b) DLT is typically evaluated within a short period whereas RT induced normal tissue complications may happen long after the follow-up (e.g. late-onset toxicity) and (c) the limited sample size (10–30) for phase I trial may be insufficient to provide an accurate estimate for toxicity. Consequently, for a phase II trial for RT using survival outcome, it is reasonable to treat time-to-disease progression and time-to-normal tissue complications as co-primary endpoints in a single trial. Moreover, for most phase II cancer oncology trials, if a patient experiences either disease progression or normal tissue complications, he/she should be treated off the protocol for ethical consideration. Since only the first event is observable, the competing risk issue arises.
Most phase II trial designs assume population homogeneity and either assign patients to a single treatment arm (Phase IIA) or randomize them to receive different treatments (Phase IIB). The randomization scheme is typically independent of patients’ personalized information, disconnected from clinical practice. For RT, recent research has revealed that patients’ responses can be remarkably different due to heterogeneous radiation susceptibility status. 9 Specifically, while some radiation-sensitive (SE) patients may yield desirable performances at a relatively low RT dose, some radiation-resistant (RE) patients require a very high RT dose to control disease.10–12 Studies in stereotactic body RT showed that a very high dose is required to reach at least 90% tumor control for RE patients with stage I non-small cell lung cancers (NSCLCs). 13 Single-institution studies and secondary analysis of Radiation Therapy Oncology Group (RTOG) trials also showed that increasing the RT dose improved local control and survival for RE patients. 14 However, as demonstrated in the RTOG 0617 trial, where a high dose arm has poorer survival than the standard dose arm in treating the SE patients, a high dose will harm the SE patients because it can induce severe and irreversible normal tissue complications. 15 Hence, a precision design is needed to (a) handle the competing risk co-primary survival endpoints (time to disease progression and time to normal tissue complications) and (b) incorporate each patient’s radiation susceptibility status (RE and SE) into radiation dose assignment and evaluation procedures.
Our study is motivated by a phase II clinical trial conducted at the Department of Radiation Oncology, Indiana University Melvin and Bren Simon Comprehensive Cancer Center. This trial aims to evaluate the PFS and monitor the normal tissue complications for stage-III NSCLC patients receiving different doses of stereotactic body RT. A total of 92 patients will be enrolled and randomized into the trial. Patients will be classified into SE and RE subgroups, using a well-established ERCC1/2 SNP signature.13,14 ERCC1/2 genes are well known for repairing the ultraviolet-induced DNA damage through the nucleotide excision repair pathway. 16 Studies also showed that they are involved in DNA repairs for ionizing radiation-induced damage. 17 There are three RT doses for consideration, referred to as the low dose (62 Gy in 2 Gy/fraction), the standard dose (74 Gy in 2 Gy/fraction), and the high dose (82 Gy in 2 Gy/fraction). Only the first two will be considered for the SE patients, and the last two for the RE patients. Each patient will be followed for six months to assess PFS. If any patient in the trial has experienced either disease progression or normal tissue complications, he/she will be treated by a second-line treatment off the protocol.
In this paper, we propose a Bayesian adaptive biomarker stratified phase II randomized clinical trial design fitting the requirement of the motivating trial. As illustrated in Figure 1, we use a proportional hazard regression model to characterize the association between the time-to-event, RT dose, and radiation susceptibility status. We treat disease progression and normal tissue complications as cause-specific events and use the cause-specific hazard competing risk model to link these two events. We construct a utility function to measure the risk-benefit tradeoff between the competing risk outcomes. Stratified by the radiation susceptibility status, we develop a response-adaptive randomization scheme. More patients will be randomized to the RT dose reporting more favorable response outcomes in the posterior mean utility estimates. A subgroup-specific RT dose will be selected for SE and RE patients separately at the end of the trial.
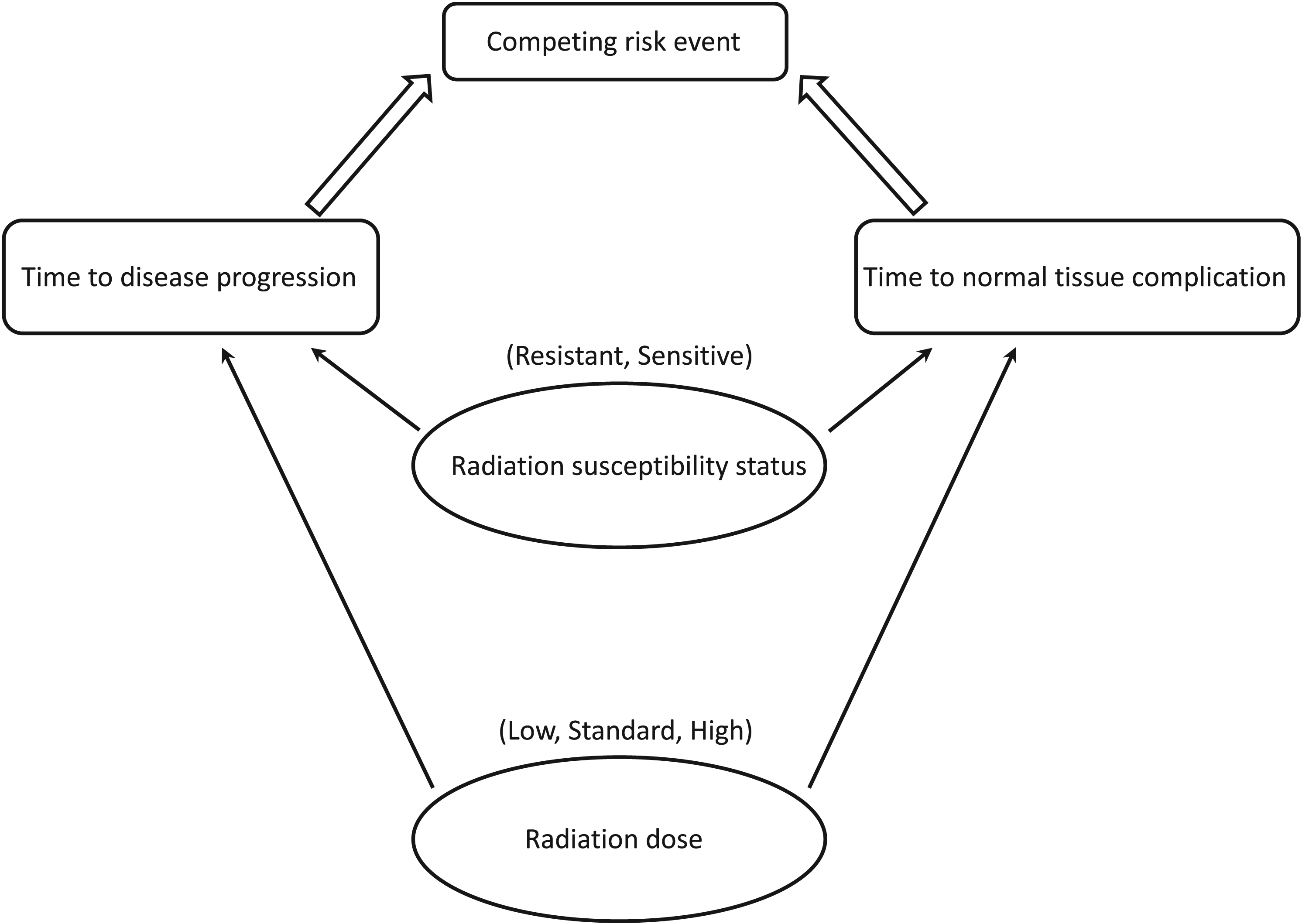
Illustration of the proposed competing risk model for cause-specific events.
Numerous phase II clinical trial designs have been proposed. Frequentist designs includes the Simon’s two-stage design 18 and its extensions.19–24 A lot of Bayesian adaptive phase II designs have also been developed using posterior probability, predictive probabilities, and Bayes factors for both single-arm trials25–29 and randomized trials.30–34 There are also adaptive designs developed for biomarker-guided phase II clinical trials, such as the tandem two-stage design, 35 sequential enrichment design, 36 parallel two-stage design37,38 and its extension, 39 and the Bayesian order constrained adaptive design. 40 However, all the existing biomarker-guided designs are only for short-term binary efficacy outcomes. To the best of our knowledge, the design proposed in this paper represents the first precision phase II clinical trial design dealing with competing risk survival outcomes.
We have proposed a Bayesian adaptive phase I/II design for competing risk outcomes. 41 The differences between the Zhang et al. 41 ’s design and this new design are (a) the previous design is for phase I dose-finding trials whereas the new design is for randomized phase II trials; (b) the previous design treats the competing risk data as an ordinal outcome and develops a Bayesian data augmentation method to impute the late-onset outcomes whereas this new design treats the competing risk data as survival outcome and uses the cause-specific hazard approach to model the competing risk survival data; (c) the previous design assumes population homogeneity whereas this new design incorporates each patient’s biomarker information; and (d) the previous design is mainly used for immunotherapies and targeted therapies whereas the new design is developed explicitly for RT. In addition, Biard et al. 42 recently proposed another phase I/II design dealing with competing risk outcomes. However, similar to our previous phase I/II design, this design is only for dose-finding trials. It cannot be directly used for a phase II trial or incorporate biomarker information.
The remainder of this paper is organized as follows. In Section 2, we describe the probability model. In Section 3, we present the biomarker stratified design. In Section 4, we investigate the operating characteristics of the proposed design through numerical studies. In Section 5, we propose an illustrative trial example. We provide concluding remarks in Section 6.
We first develop the competing risk probability model for survival outcomes. For the
We use the cause-specific hazard approach to model the competing risk outcomes. Let
There are many ways to specify
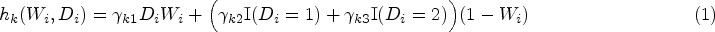
After specifying
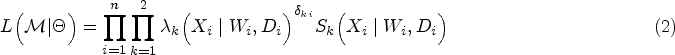
We propose to estimate
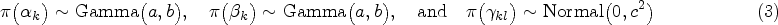
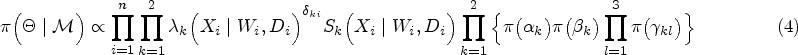
We propose the phase II clinical trial design based on the aforementioned probability model (4). The proposed design aims to evaluate the overall risk-benefit profile for each RT dose, randomize patients to receive more desirable RT doses based on patients’ radiation susceptibility statuses, and select the best subgroup-specific RT dose. Toward these goals, we need a tradeoff measurement to compromise two cause-specific events (disease progression and normal tissue complications). We propose to use a utility function to measure each patient’s survival benefit, which is a function of the RT dose given the radiation susceptibility status. The utility function should consider the event-happening time point because the later event is preferable.
Assuming that each patient will be followed in a time interval
An example of the desirability weights for the utility function with the cutoff value
.

An example of the desirability weights for the utility function with the cutoff value
We note that the utility function is very general, and the desirability weights can be easily tailored to each trial’s specific requirement. For example, if the event’s time point is unimportant, we can specify 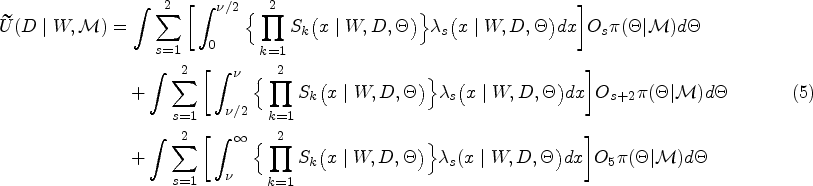
In addition to the utility function, we construct two admissible sets to safeguard the patients in the trial. Developing these admissible sets aims to avoid treating patients at overly toxic or less efficacious RT doses. To achieve this goal, we propose continuously monitoring cumulative incidence rates (CIRs) for disease progression and normal tissue complications events. The CIR is the probability that a cause-specific event occurs first within 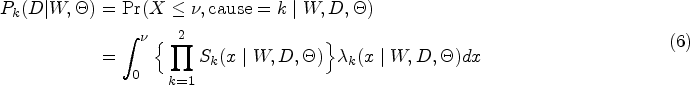
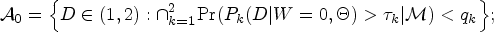
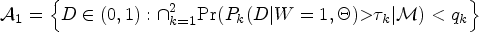
Our proposed biomarker stratified design starts by equally randomizing first Construct the admissible sets If If We repeat steps 1–3 until the trial is early terminated or the maximum sample size is reached.
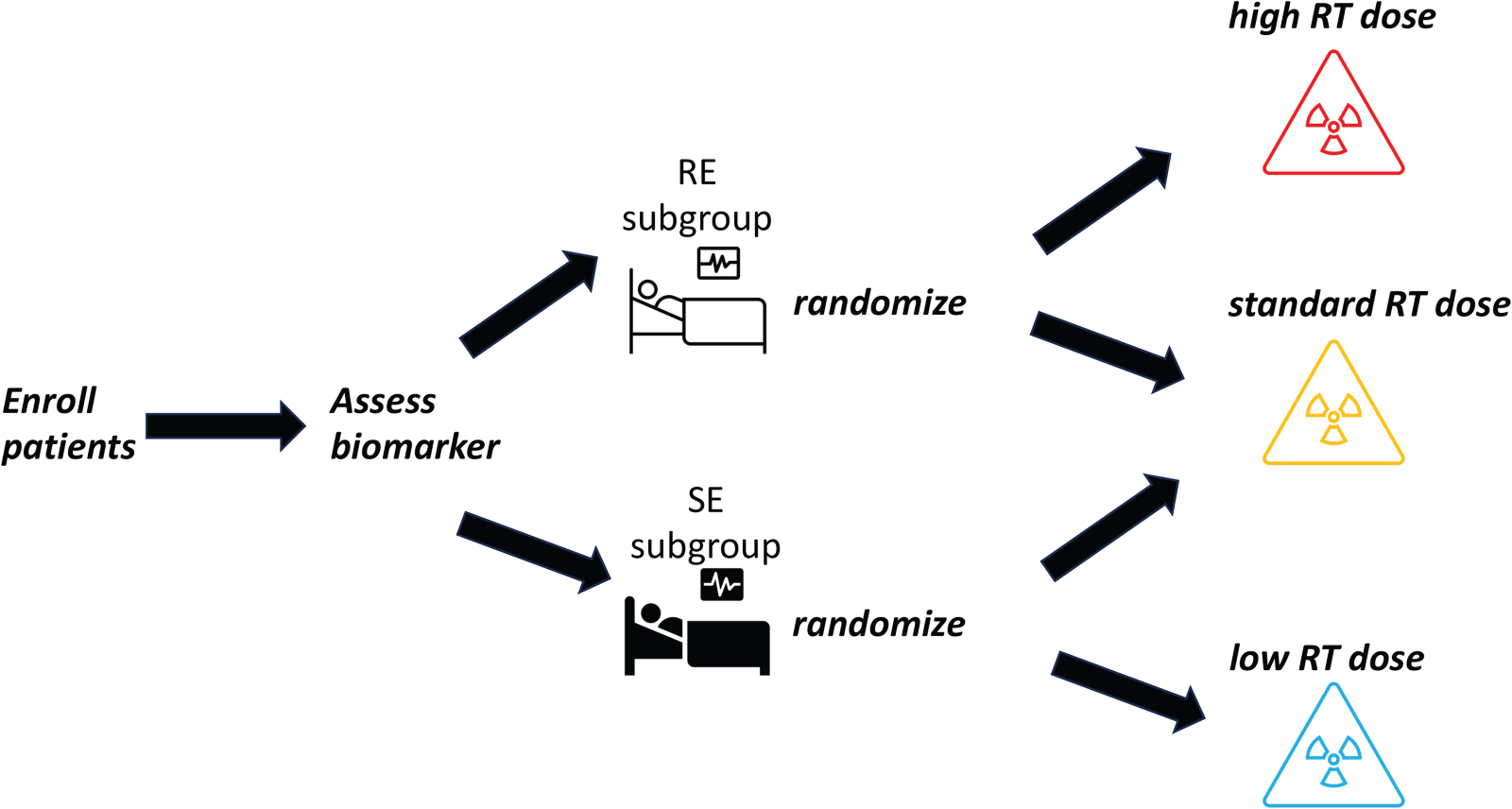
The diagram of the biomarker stratified design.
At the end of the trial, if there is only one RT dose remaining in the admissible set for either RE or SE patients, that RT dose is recommended as the subgroup-specific RT dose for the corresponding radiation susceptibility status subgroup. Otherwise, let us define
As an essential step to apply the proposed design to the motivating NSCLC trial, we evaluate the operating characteristics of the design through numerical studies. Reporting operating characteristics is often required in trial protocols when a new design is involved.
We specified that each cohort consisted of 5 patients and enrolled 4 cohorts for equal randomization and 16 additional cohorts for response-adaptive randomization. So the maximum sample size of the trial was 100. As a Bayesian design, the trial’s sample size is determined through simulation calibration. In particular, We first enumerate all the practically feasible settings of the sample sizes ranging from 50 to 150. We then simulate 5,000 independent trials using the proposed design to obtain the design’s empirical statistical power, and finally select the sample size based on the statistical power. In this simulation study, we select 100 because it is the minimal sample size, achieving at least 50% power across all the scenarios under consideration.
The radiation susceptibility status
We compared the proposed design with two alternative designs. The first design ignored the competing risk issue and modeled the time to disease progression and time to normal tissue complications events separately. We refer to this design as the “separate” design. The second design used equal randomization instead of response-adaptive randomization, and we refer to this design as the “ER” design. We refer to the proposed design as the “AR” (adaptive randomization) design. We consider eight scenarios for the numerical studies. Under each scenario, we simulated 5000 trials. Details of Scenarios 1-8 are given as follows:
In Scenario 1, the amount of decrease in CIR for disease progression is equal to the increase in CIR for normal tissue complications. In Scenario 2, the amount of CIR decrease for disease progression is higher than the amount increase in CIR for normal tissue complications. In Scenario 3, only the standard RT dose is admissible for the RE patients, and none of the RT doses are admissible for the SE patients. In Scenario 4, only the standard RT dose is admissible for the RE patients, and only the low RT dose is admissible for the SE patients. In Scenario 5, both the standard and high RT doses are admissible for the RE patients. The amount of decrease in CIR for disease progression is larger than the amount of increase in CIR for normal tissue complications. Only the standard RT dose is admissible for the SE patients. In Scenario 6, only the high RT dose is admissible for the RE patients. The low and standard RT doses are admissible for the SE patients, and the amount of CIR decrease for disease progression is less than the amount of CIR increase for normal tissue complications. In Scenario 7, both the standard and high RT doses are admissible for the RE patients. The decrease in CIR for disease progression is equal to the increase in CIR for normal tissue complications. Only the standard RT dose is admissible for the SE patients. In Scenario 8, all doses for SE and RE patients are admissible. The amount of decrease in CIR for disease progression is less than the increase in CIR for normal tissue complications.
Table 2 summarized the operating characteristics of the three designs under investigation, including the RT dose selection probability, the number of patients treated, and early stopping percentages, all stratified by the radiation susceptibility status. The RT dose selection probability indicates the benefit for further patients outside the trial, and the number of patients treated indicates the benefit for current patients.
The results of numerical studies based on 5000 simulated trials.
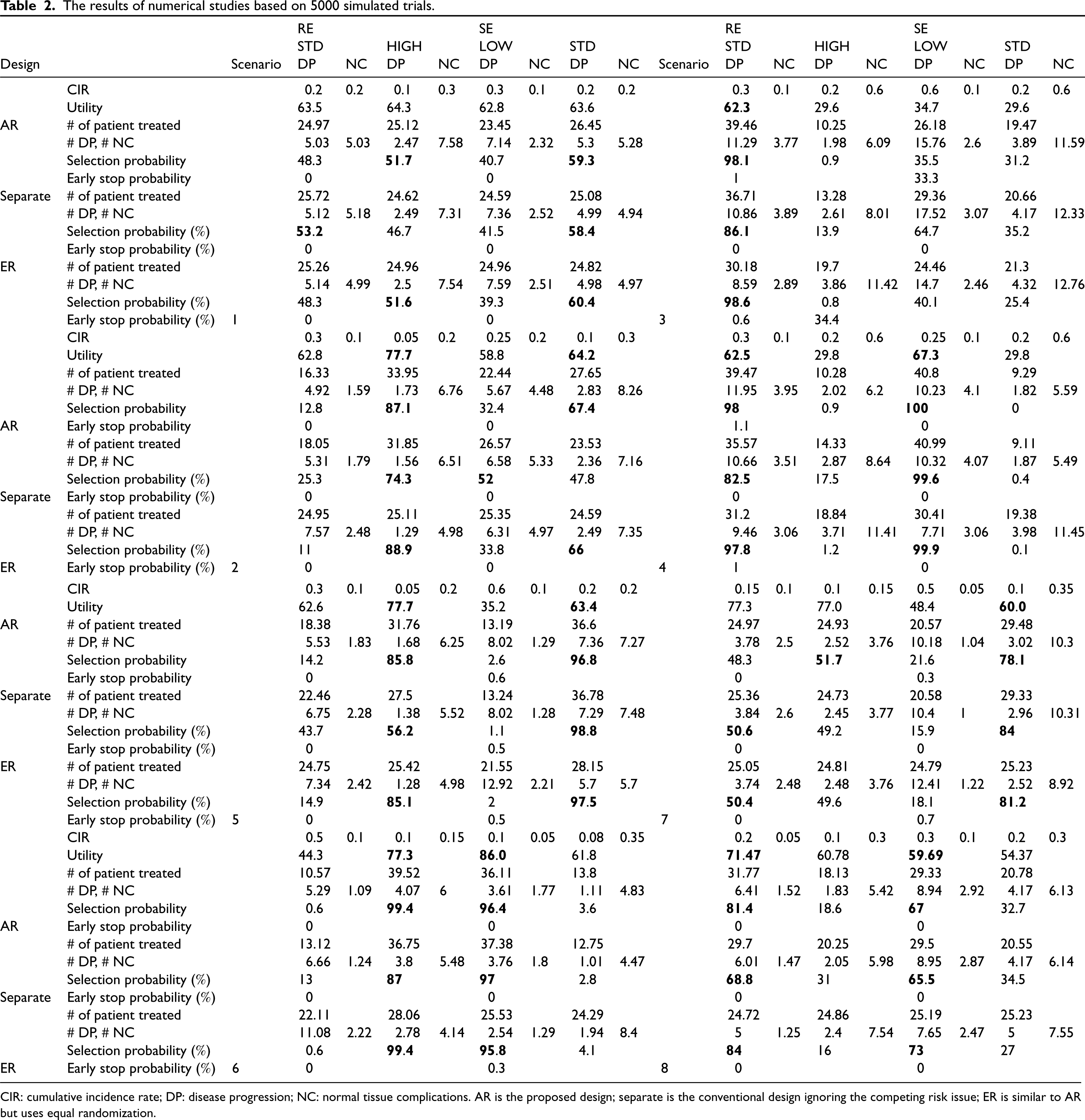
The results of numerical studies based on 5000 simulated trials.
CIR: cumulative incidence rate; DP: disease progression; NC: normal tissue complications. AR is the proposed design; separate is the conventional design ignoring the competing risk issue; ER is similar to AR but uses equal randomization.
In Scenario 1, the selection probabilities for the proposed AR and ER designs were comparable. The utility values of the RT doses were close within each subgroup under this scenario. Both designs yielded higher probabilities in selecting the RT doses with higher utility values. However, the Separate design, which ignored the competing risk issue, was preferable to the RT doses with fewer utility values. The total number of patients in the trial is identical for all the designs. However, due to the advantage of response-adaptive randomization, the AR design assigned more SE patients to the standard RT dose than the other two designs. The early stopping percentages were close to 0 for all the designs because all the RT doses were in the admissible set.
In Scenario 2, the AR and ER designs exhibited desirable subgroup-specific RT dose selection probabilities, significantly outperforming the Separate design. In addition, the AR design was the most ethical regarding patient allocation as it allocated the most patients to the true optimal RT dose, maximizing the survival benefit. Indeed, let us consider the ratio of patients assigned to the optimal RT dose to those assigned to the non-optimal RT dose (optimal/non-optimal ratio) as a measurement for the individual ethics of the design. The ratio was 2.07 for the AR design, 1.76 for the Separate design, and further dropped to 1.01 for ER because of equal randomization.
In Scenario 3, all designs had overwhelmingly high correct subgroup-specific RT dose selection probabilities for the RE patients. The AR and Separate designs outperformed the ER designs regarding patients allocation. There were no admissible RT doses for the SE patients. Compared with the Separate design, the AR and ER designs yielded around
In Scenario 4, since only one RT dose is admissible for both subgroups, all three designs had high correct RT dose selection probabilities. However, the AR and ER designs yielded at least
In summary, the proposed AR and ER designs surpassed the Separate design regarding correct subgroup-specific RT dose selection, and the improvement can be substantial. The AR design generally performed best for the patients’ allocation across all the scenarios due to the response-adaptive randomization scheme. Therefore, by jointly considering the RT dose selection and patients’ allocation, we recommended the proposed AR design being used in practice.
We performed additional simulation studies to evaluate the sensitivity of the proposed AR design to several of its design parameters. The results are summarized in the online supplemental material. The time-to-event outcomes in Table 2 were generated from the Weibull distribution. Tables S1 and S2 summarize the results using the exponential and log-logistic distributions to generate the time-to-event outcomes. The results show that the AR gives similar performances with different distributions. Therefore, the AR design is robust against the model mis-specification of the time-to-event distribution.
In addition to the desirability weights used in the main simulation study, Table S3 lists six alternative desirability weights for the sensitivity analysis. Tables S4 to S7 summarize the corresponding sensitivity analysis results. The AR design generally yields similar performances across different choices of desirability weights. Lastly, Tables S8 and S9 summarize the operating characteristics of the AR design with different cohort sizes of 2, 4 and 5. The results show that different cohort sizes yield similar design performances.
To demonstrate the practical application of the proposed AR design in clinical practice, we conduct an illustrative trial example in this section. Following the same trial settings as the numerical studies, we generate competing-risk PFS outcomes under Scenario 5 of Table 2. We compare the performance of both the proposed AR design and the conventional separate design, which ignores the competing risk issue in the trial. The complete PFS data is summarized in Figure 3. Also, we present the detailed design parameters in Table 3 for a better understanding of the trial conduct. Specifically, we provide the relevant information for the trial when the 5th and 13th cohorts of patients have been enrolled and followed for PFS, as well as the final analysis results. These include estimated utility function values, patient allocation probabilities, and the inadmissible probabilities denoted as
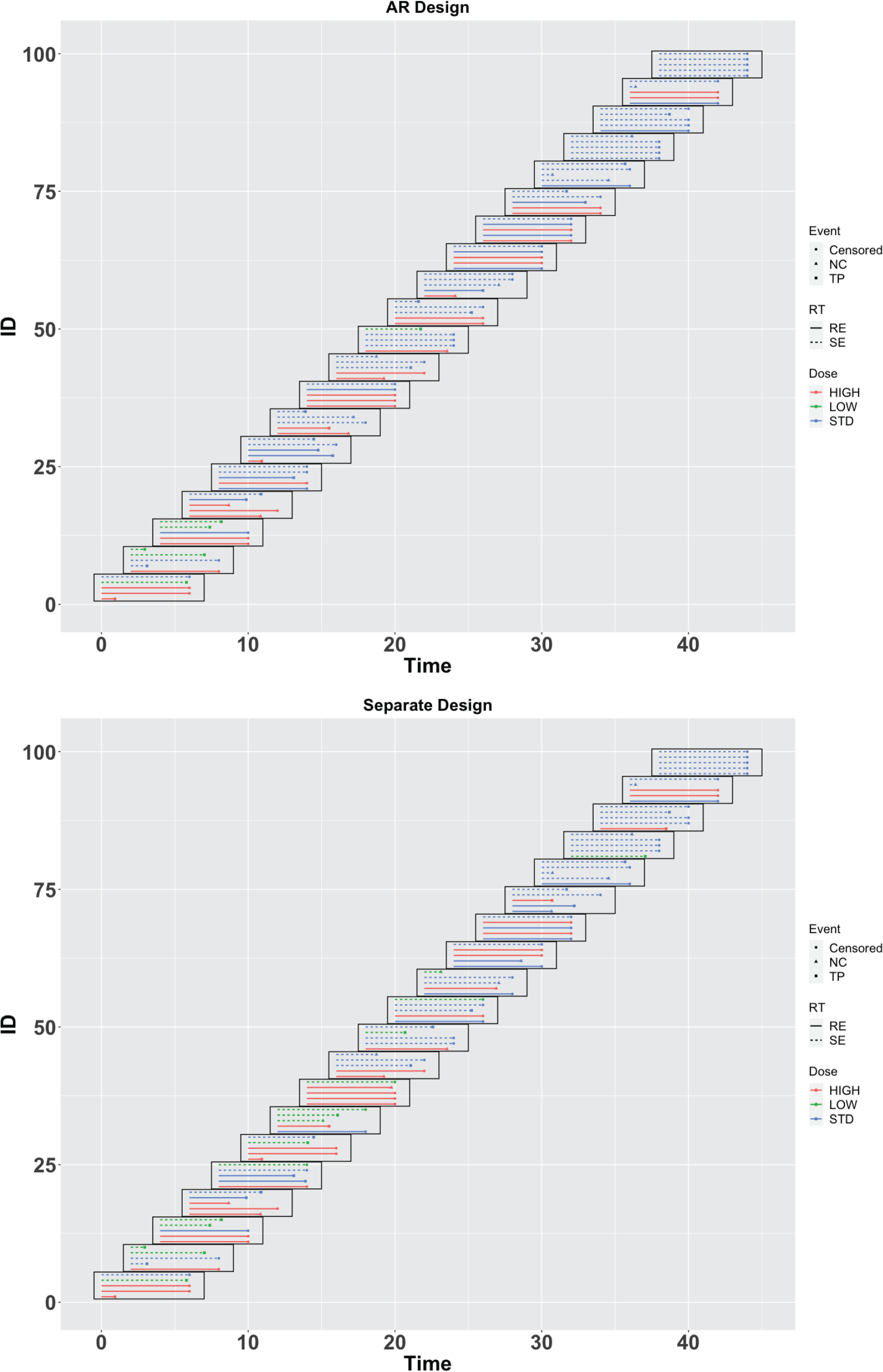
Time-to-event data of the illustrative trial.
The design parameters of the illustrative trial with Cohorts 5 and 13 and the final analysis.
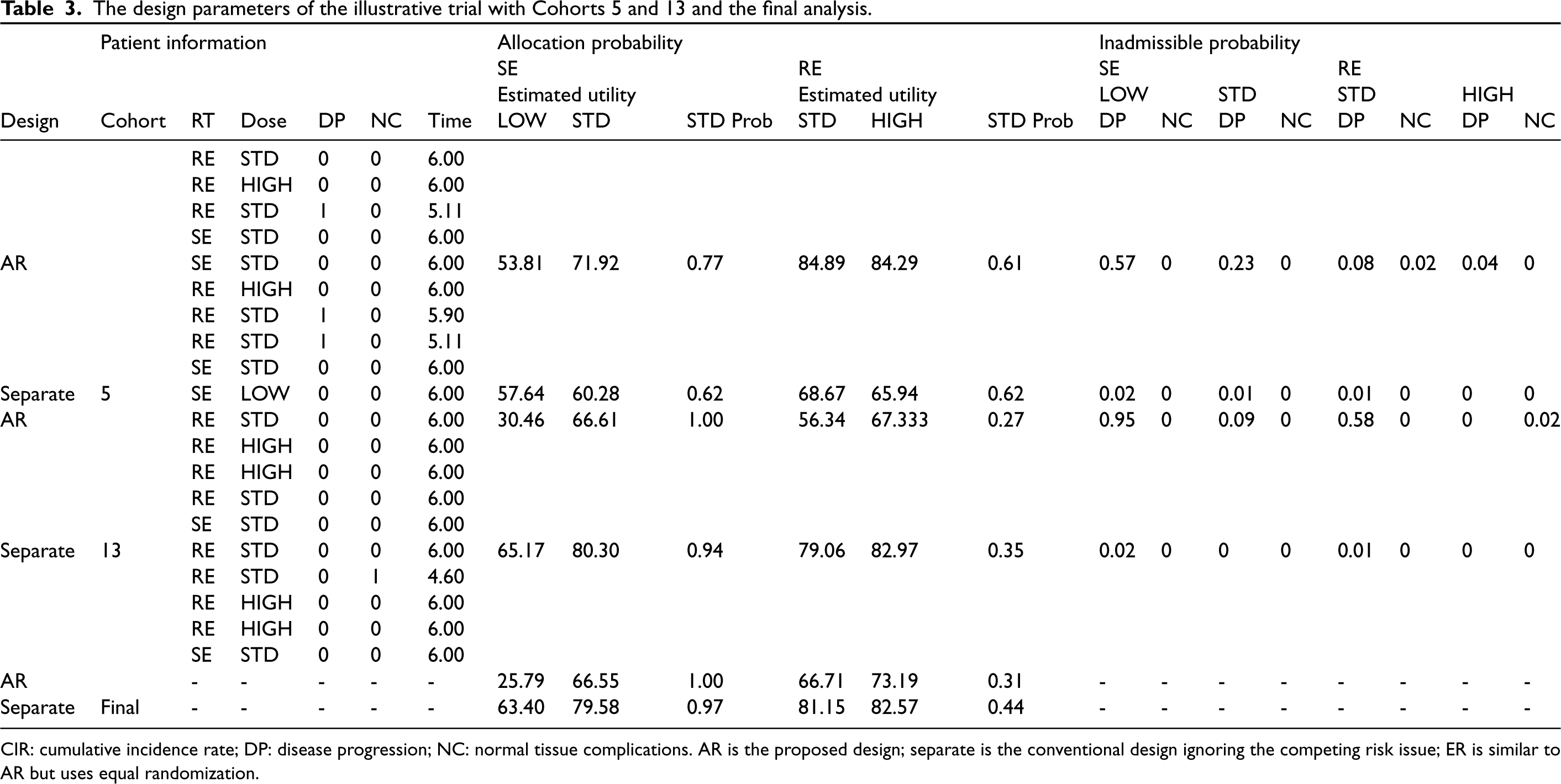
CIR: cumulative incidence rate; DP: disease progression; NC: normal tissue complications. AR is the proposed design; separate is the conventional design ignoring the competing risk issue; ER is similar to AR but uses equal randomization.
The first part of the trial involves four cohorts of patients. During this part, SE patients are equally randomized to receive either a low RT dose or the standard RT dose, while RE patients are equally randomized to receive either a high or standard RT dose. After the first four cohorts, the trial switches to a response-adaptive randomization method for subsequent cohorts of patients. Specifically, under the proposed AR design, when the 5th cohort of patients have been enrolled and followed for PFS, after applying the proposed statistical model to the accumulated data from cohort 1 to cohort 5, for the SE patients receiving the low RT dose, we get the posterior estimates for the competing risk events 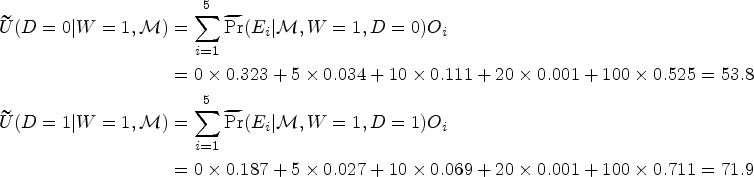
After collecting all the data, we perform the final analysis to determine the optimal RT doses under the AR design for different subgroups of patients. For the SE patients, the final posterior mean utility function estimates are 25.79 and 66.55 for the low and standard RT doses, respectively. Since the low RT dose is inadmissible, the standard RT dose should be selected for the SE patients. For the RE patients, both the standard and high RT doses are admissible, with corresponding posterior mean utility function estimates of 66.71 and 73.20, respectively. By comparing these two utility functions, the posterior probability that the standard RT dose is better than the high RT dose is 30.8%, which is less than the cut-off value of 0.5. Consequently, the high RT dose is selected as the optimal RT dose for the RE patients.
We also present the results of the illustrative trial using the conventional separate design for comparison purposes. Our observations reveal that when compared with the proposed AR design, the separate design produces significantly more biased utility function estimates. For instance, for the SE patients, the actual utility function values at the low and standard RT doses are expected to be 35.2 and 63.4, respectively. However, the separate design provides estimates of 63.4 and 79.6 for the corresponding RT doses, showing a considerable bias. Furthermore, due to these substantial biases in the estimates, the separate design consistently leads to less ethical randomization and a lower probability of selecting the true optimal RT dose when compared to the AR design. For example, in the case of the RE patients where the high RT dose is the true optimal choice, the AR design yields a probability as low as 30.8% that the standard dose is better based on the utility function. In contrast, the separate design incorrectly increases this probability to 43.7%. Based on the illustrative trial example, it is evident that the AR design exhibits superior trial operating characteristics compared to the conventional separate design. This conclusion aligns with the findings from numerical simulation studies, reaffirming the advantages of the AR design in producing more reliable and accurate results.
This paper investigates phase II trial design using survival outcomes, focusing on the RT. Both the time to disease progression and time to normal tissue complications are considered as the co-primary outcomes, so the competing risk issue arises. We built a cause-specific hazard model to solve the competing risk problem and capture the association between the time-to-event, RT dose, and radiation susceptibility status. We propose to use a utility function method to tradeoff the risk-benefit of the RT dose on the cancer cell and normal tissue, which provides an overall measurement of the survival benefit of the different RT doses. Stratified by the radiation susceptibility status, we develop a Bayesian response-adaptive randomization scheme. More patients will be randomized to the RT dose reporting more favorable response outcomes in the posterior mean utility estimates. A subgroup-specific RT dose will be selected for SE and RE patients separately at the end of the trial. Numerical studies confirm the proposed design’s desirable performances, compared with the conventional design ignoring the competing risk issue.
We construct the utility function based on two sub-intervals of the follow-up time to differentiate between early-onset and late-onset adverse events. Suppose for a particular trial, it is clinically irrelevant to make such a difference. In that case, the utility function can be easily modified for the whole follow-up time only, and the number of events should be reduced from 5 to 3. Also, the cut-off value
Supplemental Material
sj-pdf-1-smm-10.1177_09622802231215801 - Supplemental material for A Bayesian adaptive biomarker stratified phase II randomized clinical trial design for radiotherapies with competing risk survival outcomes
Supplemental material, sj-pdf-1-smm-10.1177_09622802231215801 for A Bayesian adaptive biomarker stratified phase II randomized clinical trial design for radiotherapies with competing risk survival outcomes by Jina Park, Wenjing Hu, Ick Hoon Jin, Hao Liu and Yong Zang in Statistical Methods in Medical Research
Footnotes
Acknowledgment
The authors thank two referees for their valuable comments, which substantially improved the presentation of this paper.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received the following financial support for the research, authorship and/or publication of this article: Yong Zang’s research was partially supported by NIH grants R01 GM150808, P30 CA082709, R21 CA264257, and the Ralph W. and Grace M. Showalter Research Trust award. Ick Hoon Jin’s research was partially supported by the Yonsei University Research Fund 2019-22-0210 and by Basic Science Research Program through the National Research Foundation of Korea (NRF 2020R1A2C1A01009881 and RS-2023-00217705).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.