
Abstract
Background
The number of Phase III trials that include a biomarker in design and analysis has increased due to interest in personalised medicine. For genetic mutations and other predictive biomarkers, the trial sample comprises two subgroups, one of which, say
Methods and Results
Assuming trial analysis can be completed using generalised linear models, we define and evaluate three frequentist decision rules for approval. For rule one, the significance of the average treatment effect in
Conclusions
When additional conditions are required for approval of a new treatment in a lower response subgroup, easily applied rules based on minimum effect sizes and relaxed interaction tests are available. Choice of rule is influenced by the proportion of patients sampled from the two subgroups but less so by the correlation between subgroup effects.
1 Background
Since the rise of personalised medicine, the number of Phase III trials that include a biomarker in design and analysis has increased. A biomarker has been defined as “A
In our context, we define sub-populations of patients as either biomarker positive (B+) or negative (B−). A common situation is that the treatment efficacy is a priori assumed to be better (or at least as good) in B+ compared to B− patients. For example,
a drug treatment may have been developed to target a genetic disorder defining group B+, there may be an untested clinical hypothesis of better efficacy in B+, empirical data (biological or early clinical) may indicate higher efficacy in B+.
Even if a treatment has been developed to target the biomarker of interest, some of the efficacy in B+ may obtain in B−. Depending on the situation, one may expect no or minimal efficacy in B−, that a large proportion of the efficacy in B+ is retained, or that efficacy in B− is difficult to predict even if the treatment is known to be efficacious in B+. The effect in
Although a new treatment may be most effective in the higher responding
There is a large literature on subgroup analysis in phase III trials. Much of this literature concerns post hoc exploration of a moderate to large number of subgroups using interaction tests, with issues such as data dredging and multiplicity well documented. 4 This study differs in that we are concerned with the situation where there is an overall significant effect, two pre-defined subgroups known to differ in treatment effect, and optimal rules for treatment approval in the lower responding subgroup are required.
Where hypothesis tests are applied to multiple subgroups, it is important to control family-wise error rate (FWER). 5 For two subpopulations B+ and B−, there are different multiple testing procedures that control the FWER. 6 In most applications, formal testing focuses on F and B+ using either a hierarchical approach (F followed by B+, or B+ followed by F) or by splitting type I error between parallel tests; testing of B− is rarely included in applications for regulatory approval, as power is considered to be limited.7,8 In this study, we concentrate on the situation where the intervention has statistically significant efficacy in F, with significance in the B+ group assumed to follow due to higher efficacy in this subpopulation; conditions for approval in B−are then developed and assessed. A strategy that conditions on significance in B+ rather than F is closely related mathematically and results are expected to be very similar.
Although the study is motivated by trials of drugs targeting specific genetic mutations (see Gonzalez-Martin et al. 9 for a recent example), other examples of trials with similar structures define subgroups according to age (adults and children), 10 mild and severe disease, 11 early and late stage cancers, 12 as well as other biomarkers. 13 Proposed methods should apply to any trial including two subgroups with known or suspected treatment effect inequality.
We provide a brief literature review of methods and practice in this context in section 2 before describing proposed rules for exponential family models in section 3. Conditional power of the rules is explored in section 4 and applied retrospectively to two published phase III trials in section 5, before briefly discussing implications for future trial design. A discussion completes the paper (section 6).
2 Existing literature
A review of FDA drug approvals with required biomarker testing found that biomarker negative patients were simply excluded from the majority of trials. 14 Since exclusion was often not based on clinical evidence, these patients could be denied potential benefit from novel treatments. Moreover, provided that there is a sound biological basis for some benefit in biomarker negative patients, including them may also confirm the clinical utility of the biomarker itself.
Heterogeneity within a target population was also recognised in updated EMA guidance on the investigation of subgroups in confirmatory clinical trials, published in January 2019.3 Whilst the guidance suggests that restriction of a trial population to a sub-population is justified if there are safety concerns or an anticipated lack of efficacy, it also calls for additional trials including the full breadth of the population to provide the best evidence of effect modifiers. Inclusion of the biomarker negative subgroup was highlighted as important, though it can create difficulties in analysis if the treatment effect is small or there are only a small number of such patients, resulting in low power to detect a significant treatment effect. Despite this, patients in this subgroup may still benefit from treatment, and are therefore harmed if the result is discarded for non-significance.
A 2016 review by Ondra et al. 15 found that two concepts underpin current methods for assessing subgroup effects, influence and interaction. An ‘influence’ condition sets a threshold that must be met by the treatment estimate of the subgroup of interest, whilst an interaction test sets a difference between treatment effects for two (or more) subgroups, in effect requiring that effects are sufficiently close. These methods may be used for approval of a treatment or as conditions which must be met for the subpopulation to be included in the next stage of analysis (adaptive designs). For example, Stallard et al. 16 compared different strategies for choosing which hypotheses to test in the second stage of analysis (either the full population or a subgroup, or both), which used either an influence or interaction test approach. Similarly, Matsui and Crowley 17 proposed a sequential design where in the first stage of their analysis they use superiority and futility boundaries to decide which populations go forward for further analysis. This preserves statistical power for detecting various profiles of treatment effects across the subgroups, and allows the biomarker negative population to be tested again if they do not cross the futility boundary.
Despite the development of different adaptive designs, interaction tests appear to be the main method used to assess subgroup heterogeneity. In our (unpublished) targeted systematic review of large clinical trials that carried out subgroup analyses in the New England Journal of Medicine, we found that approximately two thirds used interaction tests to decide whether there was significant treatment effect heterogeneity. Almost all other articles summarised within-subgroup effects and used significance tests with 5% type I error.
Although most of the literature rests on the frequentist paradigm, a Bayesian approach could also be considered. 18 By specifying a two-dimensional prior for efficacy in B+ and B−, one can explicitly borrow information from one subpopulation when evaluating the other. This prior should reflect the clinical plausibility of a range of differential treatment effects between the two subpopulations.
This study was partly motivated by the design of the APEX trial, which compared betrixaban with standard dose enoxaparin in medically ill patients at risk of venous thrombosis. They carried out sequential analyses, the first on a subgroup defined by the biomarker D-dimer, the second on a subgroup defined by a combination of D-dimer level and age, and the third of the full population. If any result was negative, then subsequent tests were treated as exploratory. The first subgroup analysis was just above the pre-defined threshold for statistical significance of 5% (p = 0.054), so that the subsequent subgroup analysis (elevated D-dimer level and age
In our context, we may accept some heterogeneity between subgroups, provided that there is sufficient benefit in the B− subgroup. The issue is in choosing an acceptable difference between the treatment effect in the two subgroups, or equivalently, choosing a relaxed (higher) significance level for the interaction test. On the other hand, decision rules that focus on influence rely solely on the data in the B− subgroup, but require us to pre-specify a minimum bound for the acceptance threshold. In order to avoid the need to specify either a minimum treatment effect or a more relaxed interaction level, a decision rule that does not require additional parameters may also be attractive.
The question of how to deal with approval in a limited sub-population has important implications for maximising the patients who could benefit, which is particularly important for conditions where there are few treatment options. There remains uncertainty about how to address this issue, and how different decision rules perform according to issues such as prevalence of the high responder subgroup in the population and in the trial. We outline a simple strategy to choose appropriate and efficient decision rules in a frequentist framework.
3 Methods
3.1 Generalised linear model and subgroup notation
In practice, phase III clinical trials that have a biomarker-treatment interaction are analysed using linear, generalised linear or survival regression models. We restrict attention in this paper to the wide range of trial outcomes that have Normal, Binomial or Poisson distributions and review the general framework here, defining estimands of interest, estimators and statistics.
Generalised linear models that describe different treatment effects in the two subgroups have a linear predictor of the form
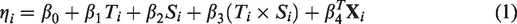
For the exponential family of distributions, the expected response and the linear predictor are connected through the link function
We define the
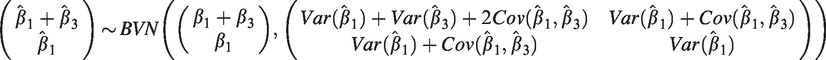
For inference for each group separately, we define Z-statistics
3.2 Likelihood assuming no correlation between
and
In order to gain insight into the contribution of π, the proportion of trial patients drawn from the high-responding subgroup
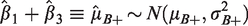
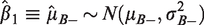
Specifically, for the response Yi, Normal distribution-Identity link Binomial distribution-Logit link Poisson distribution-Log link
We note that in all three cases the variance includes the term
3.2.1 Making inferences in the full population in the general case.
We write the full population treatment effect as

That is, the estimand for the full population is a weighted average of the subgroup specific estimands
Since
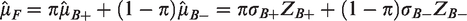
If ρ is the correlation between the estimands, the Z-statistic for the full population is
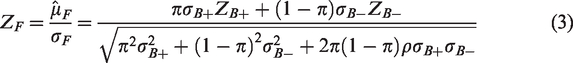
If there are no additional covariates in the model (
Alternatively, if
As an aside, we note that the correlation between the statistics
3.3 Proposed rules for approval of the drug in
3.3.1 Sequential testing and conditional power.
We define and evaluate three proposed rules for approval in the lower response population
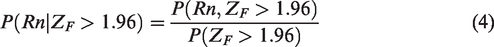
(As an aside, for binary data where the analysis adjustment for baseline covariates is not required, this conditional probability can be calculated in closed form.)
The denominator does not depend on the form of any proposed rule and is given by the observed statistic in the full population
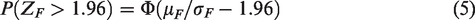
Note that the right-hand side is a function of three location parameters
We now consider conditional power of three classes of decision rule for approval of the drug in
Proposed rules for approval in the lower treatment response subgroup

Rules 1 and 2 are different types of influence rule, whilst Rule 3 is an interaction test. The algebra for calculating the conditional power of each rule is given in full in Appendix 1.
3.4 Rule 1: the statistic
exceeds a pre-defined threshold L
For the treatment to be acceptable in the
For rule 1, conditional power is given by the expression
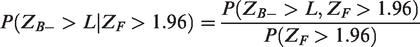
For the numerator 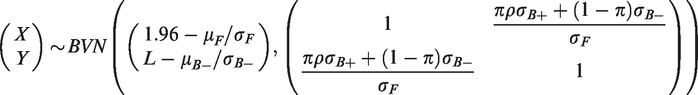
Again, the numerator of the conditional power is a function of three location and three variance parameters
3.5 Rule 2: The
data should increase statistical significance
For interventions with a low adverse event profile, approval may be acceptable provided that the data in
Making the transformations, 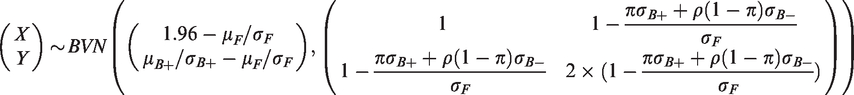
For the conditional power numerator, we calculate
3.6 Rule 3: No significant subgroup-treatment interaction at αI level
When a range of subgroup effects are explored (often post hoc), it is customary to perform interaction tests to identify specific subgroups for which the treatment appears particularly effective/ineffective for further investigation. From our targeted systematic review of literature, the type I error rate αI is almost invariably set to 5%, with no adjustment for multiplicity. Our objective here is quite different; specifically we use αI as a measure of how confident we are that the two subgroups have different treatment effects, in order to decide whether approval in
For this rule, the numerator is 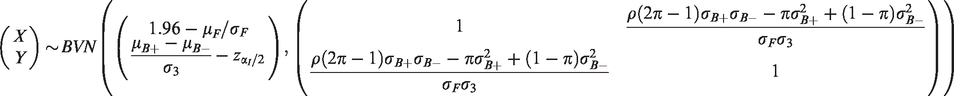
Again, the numerator for conditional power is
3.7 Illustration of proposed rules
Figure 1 illustrates the proposed rules for the (hypothetical) case of independent normally distributed estimands for the two subgroups (on the scale of analysis e.g. linear, log, logistic). The statistics
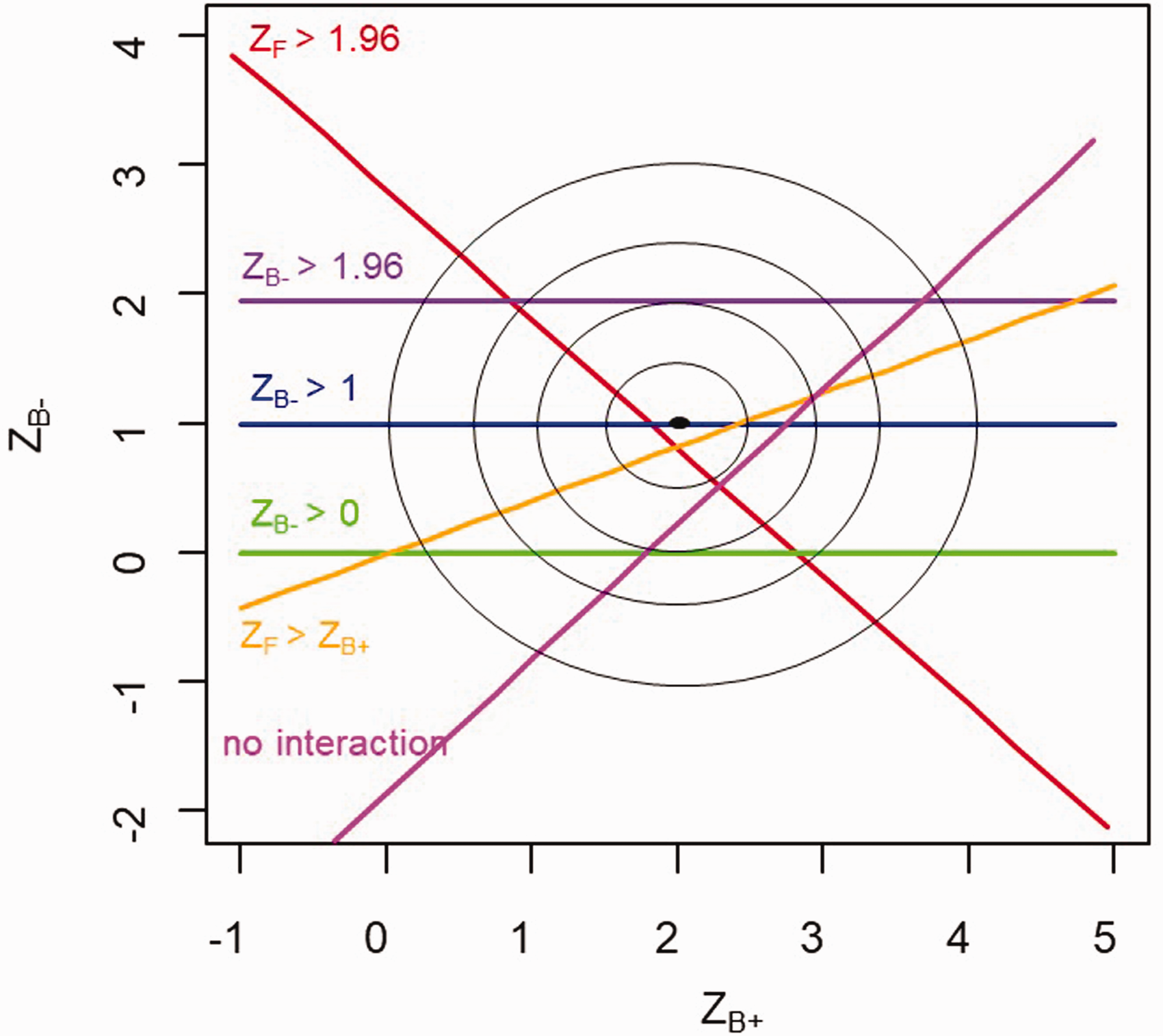
Illustration of the proposed rules in the
In general, the conditional power for each of these three rules is given by the proportion of the density of (X, Y) that is consistent with the condition
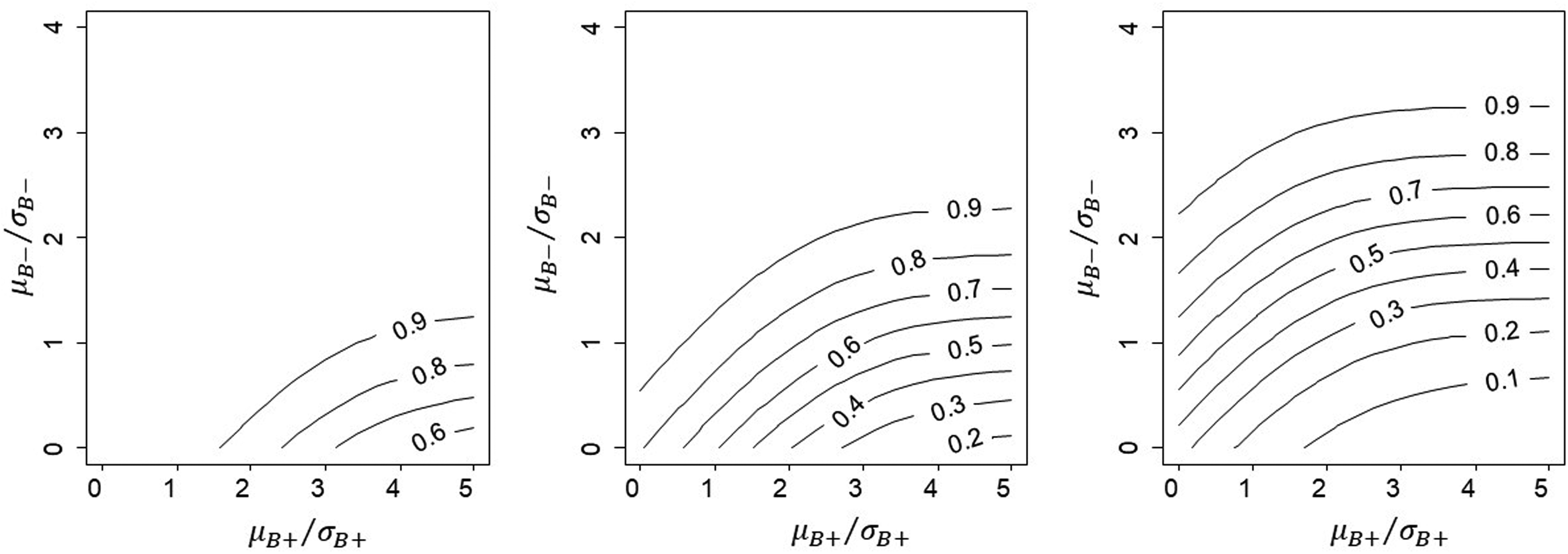
Illustration of conditional power for the case where
Because
Similar patterns can be found for Rules 2 and 3.
4 Results
4.1 Comparison of conditional power for the proposed rules
The conditional power for all three rules depends on the relative treatment effects in the two subgroups, which is driven by the biological mechanisms of the treatment. Above this, we explore how the proportion sampled from each sub-population π and
4.2 Comparison of conditional power for proposed rules when
and ρ = 0
The top row of Figure 3 shows the relationship between the subgroup-specific statistics for treatment effect and conditional power in the simple case of equal standard errors, equal numbers in the two subgroups and independent estimands. For any value of
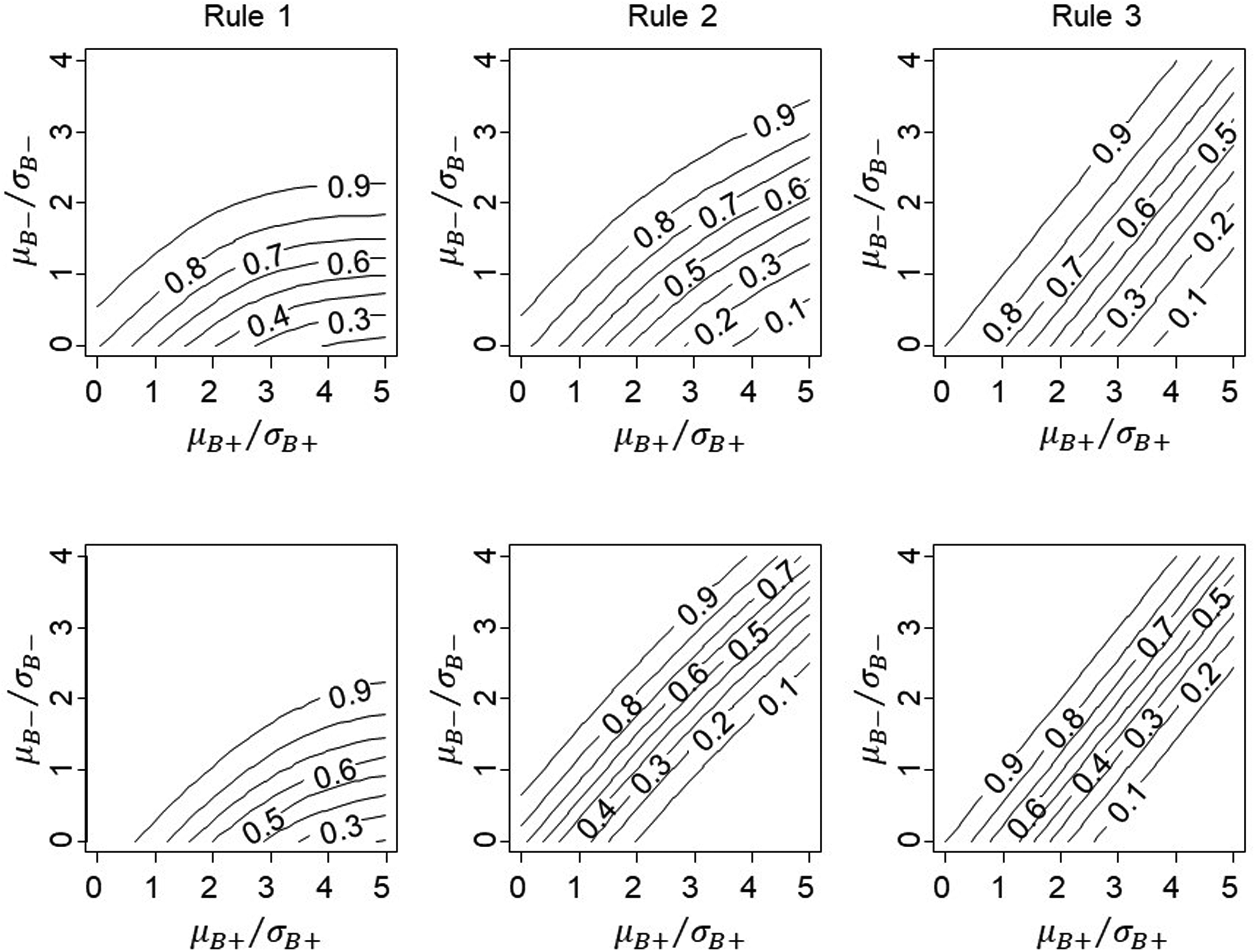
Conditional power for proposed rules with correlation between
For this illustration, we set L = 1 for Rule 1 and one-sided significance of the interaction to 0.1 for Rule 3. Changing these thresholds will result in a shift in the contour plots, whilst Rule 2 does not require specification of an additional parameter and is fixed. It is possible to closely align the three rules by choosing appropriate values for L and the interaction type I error if necessary.
4.3 Effect of correlation between estimands when
and
ρ = 0.5
Including additional covariates
We note here that, if the two estimands are independent (ρ = 0) and arise from normally distributed data with common sampling variance in the two subgroups, then conditional and unconditional power for Rule 3 are identical, since
4.4 Effect of relative subgroup size when sampling variance is homoscedastic across subgroups and ρ = 0.5
We illustrate the influence of subgroup size on conditional power for the case where sampling variance is homoscedastic across biomarker subgroups. This will occur for normally distributed outcomes with the same sampling variance
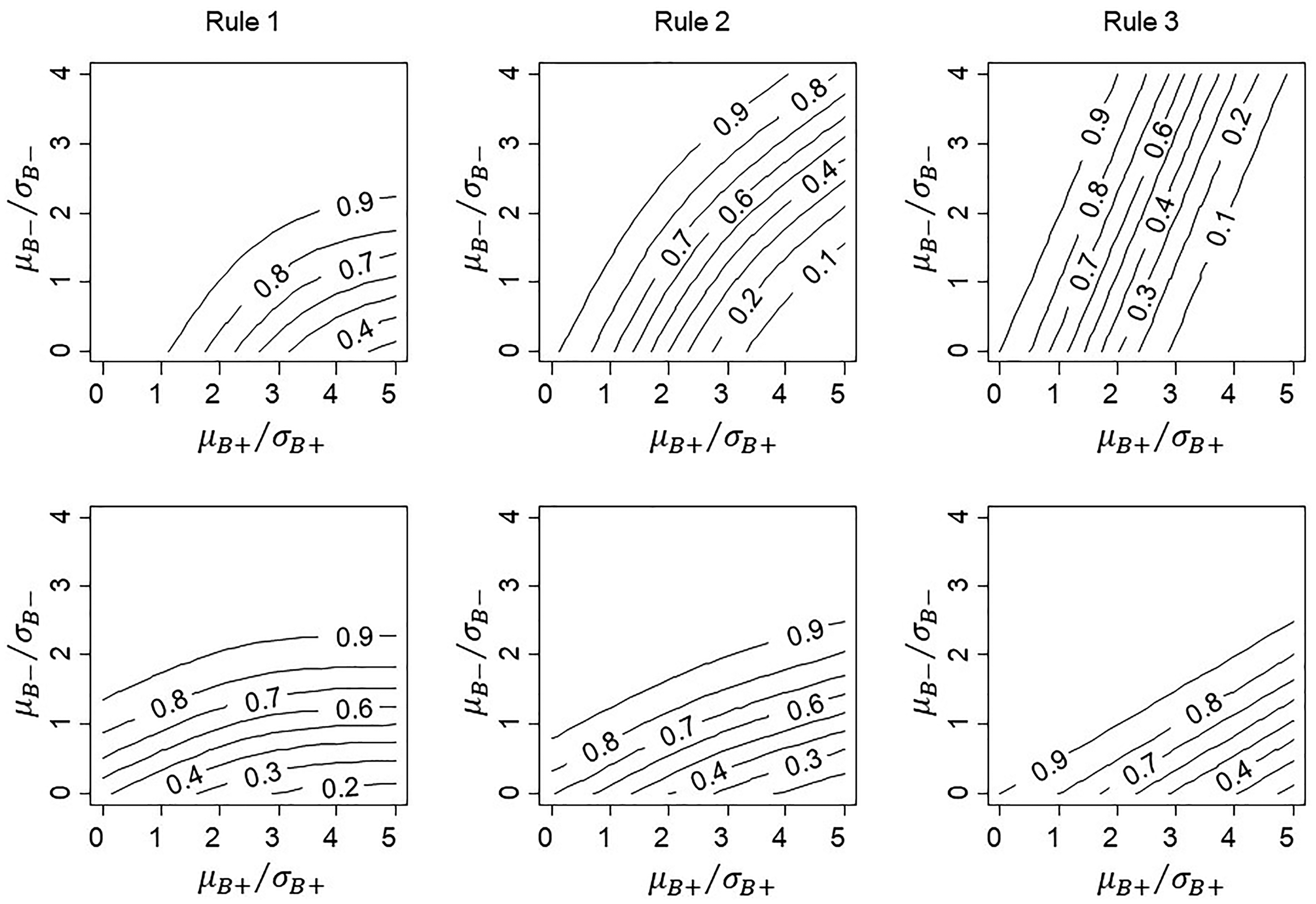
Conditional power for proposed rules with 20% of patients in subgroup
If the trial sample contains a high proportion drawn from the
To provide further insight into the effect of relative sample size of the two subgroups, we plot conditional power against
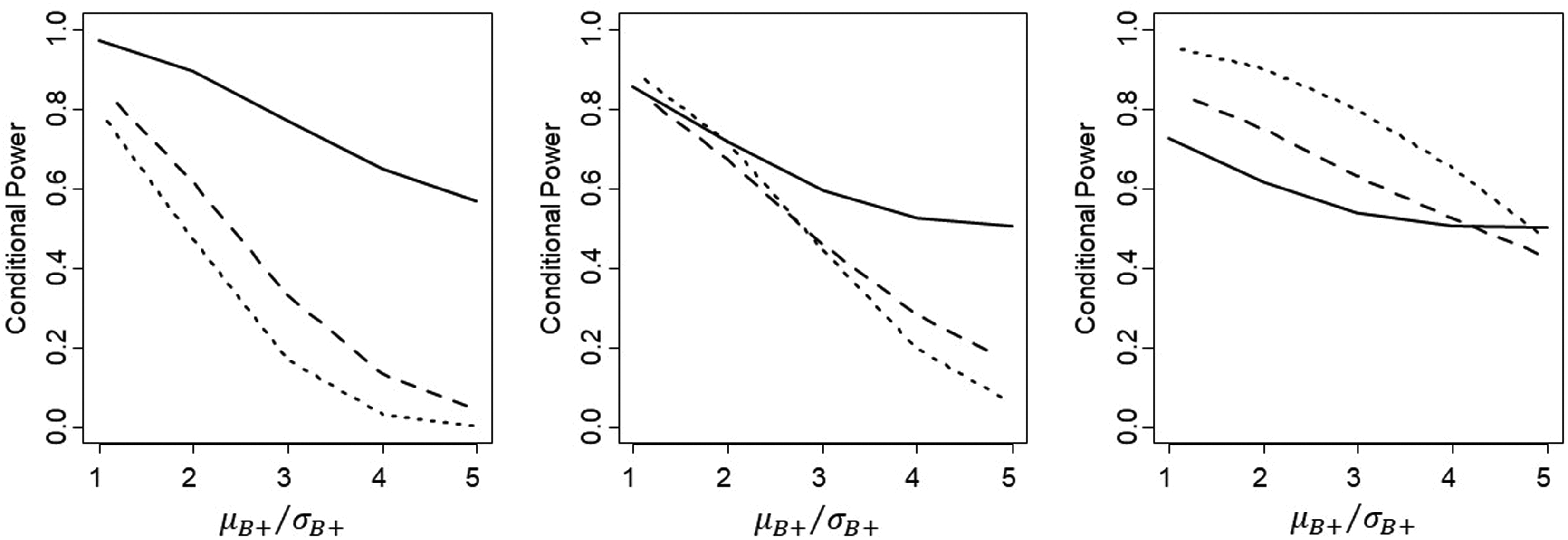
Comparison of conditional power for proposed Rule 1(solid line), Rule 2 (dashed line) and Rule 3 (dotted line), with the proportion of patients sampled from
5 Illustrative applications
In order to illustrate how these rules might be used in practice, we retrospectively apply them to two completed phase III trials: a small cardiac surgery trial of 352 patients and equal size subgroups, 19 and a much larger stroke trial (n = 7513) which evaluated betrixaban. 13
5.1 AMAZE trial in cardiac surgery
AMAZE was a cardiac surgical trial in patients with atrial fibrillation (rapid/irregular heart rhythm).
19
This multi-centre RCT randomised 352 patients 1:1 to a technique called ablation in addition to planned surgery, or to planned surgery alone (control arm). The primary outcome was return to sinus rhythm at one year post-surgery (binary). Although not part of the intervention, at the discretion of the operating surgeon 150/352 (
Treatment effect estimates and Z-statistics for patients with the LAA left intact (
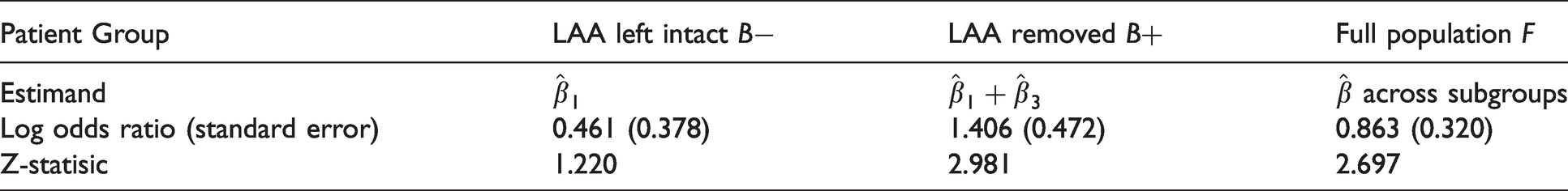
We estimate conditional power for approval of ablation in
5.2 APEX trial in patients at high risk of stroke
Recall that one of the trials motivating this study compared treatment with the anticoagulant betrixaban with standard treatment of enoxaparin amongst hospitalised medically ill patients. 13
The primary outcome was a composite of clinical events caused by blood clotting (deep vein thrombosis, non-fatal pulmonary embolism or death from thromboembolism) up to day 42 post-randomisation.
The planned analysis took a sequential testing approach, but rather than starting with the full trial population, the order of testing began with a subgroup with a high chance of treatment response (but smaller treatment effect), followed by testing in two other pre-specified, progressively inclusive cohorts as follows:
Compare treatment arms in patients with elevated D-dimer level (for illustration can be considered Compare treatment arms in patients with elevated D-dimer or age Compare treatment arms in all enrolled patients (full population, n = 7513).
If any test was negative, all subsequent tests were reported as exploratory. We provide selected results from the original trial publication in Table 3.
Treatment effect estimates and Z-statistics for patients with the elevated
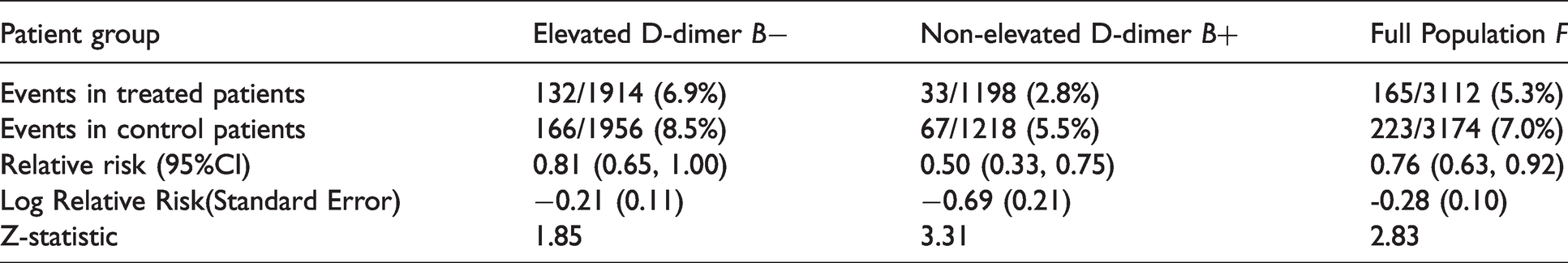
As the table shows, the first analysis including Rule 1: Rule 2 was also not met because group There was a significant interaction between subgroup and treatment (one-sided p = 0.0184) so that this trial also fails Rule 3 – the conditional power was 43%.
In summary, using our proposed sequential testing procedure, betrixaban would be recommended for treatment in elevated
5.3 Implications for trial design
In this context, investigators define and document decision rules for the primary trial analysis during the design stage. Our proposal is that, should a separate decision on approval of a subgroup be required, then a decision rule should be agreed in discussion with regulators or other appropriate decision makers during the design phase. Our evaluation of three potential rules illustrates how to investigate the efficiency of different rules, although parameter inputs will be specific to each trial and will depend on available information around potential efficacy.
Although our rules rely on Z-statistics for hypothesis tests, it is more usual to work with potential treatment effects and their standard deviations when designing a trial. Empirical estimates of variation in the primary outcome are typically available, particularly for the control arm of the trial. This may be a standard deviation for a continuous outcome, or the baseline risk of an event for patients receiving the current best treatment. Given these estimates, the sample size required for an overall significant treatment effect, the proposed sampling proportion, and the Rule 1 threshold L can be decided to ensure that the treatment effect in subgroup
The stages of design are as follows:
Using initial estimates of design parameters, including the sampling proportion π and correlation ρ, calculate the power of the test for the expected value of the treatment effect in the full trial population. Choose a decision rule for recommending treatment in Given the sampling proportion π and the expected treatment effect sizes in the two sub-populations, calculate the power of your preferred rule, conditional on a significant overall test. Calculate the power of the sequential testing strategy as the product of conditional and unconditional power in 1 and 3.
In practice the final power calculations will require an iterative process between calculation and elicitation of expert clinical knowledge of treatment effects and associated variance components, finalised in discussion with regulators or other decision makers.
6 Discussion
6.1 Overview of results
Frequentist rules to assess whether approval of a new treatment should be accepted in a lower response subgroup, conditional on a significant effect overall, have been developed and evaluated. Approval based solely on a significant overall test may be unacceptable if there are severe side effects and/or if the subgroup drawn from the low response population is under-represented due to enrichment sampling. Rules are based either on measures of influence, such as the size of the effect in this subgroup, or the increase in significance due to inclusion of the subgroup, or on the difference in effect size between the groups (interaction). When choosing a rule during trial design, as well as specifying estimates of the expected outcomes and their variance components, investigators must either take a random sample from the full population, in which case the trial will represent clinical practice, or decide the proportion of patients to be sampled from each sub-population. Using conditional power as a measure of efficiency, the proportion of patients drawn from each sub-population had a large impact, but correlation between the groups induced by covariate adjustment was less important. For all rules, conditional power decreased as μ B + /σ B + increased for fixed μ B − /σ B − .
6.2 Discussion of individual rules
After ensuring that
Rule 2 (
Rule 3 uses an interaction test with a relaxed significance level to recommend approval in the
6.3 Regulator input
In practice, acceptability of these approaches will depend on regulators (for drug trials) or commissioners (for academic trials). Since the conditional power of the rules depends crucially on the values chosen for the parameters L and αI, as well as patient sampling, prevalence of high/low responders and analysis methods, early engagement with regulators/commissioners to discuss these decision rules is worthwhile. Discussions also need to consider potential harms (side effects), in order to set realistic and acceptable targets for efficacy. In practice, investigators/sponsors will be required to pre-specify and document these decision rules in discussion with regulators.
6.4 Strength, weaknesses and future research
One benefit of our proposed decision rules is that closed-form expressions for conditional power are available for continuous, binary and count outcomes (assuming known variances). This makes estimation of sample sizes relatively simple, and a wide range of scenarios can be explored during the design phase.
In our examples, we used retrospective power calculations to show the differences in conditional power for the three rules based on trial results. We stress that these calculations were provided for illustration only and we do not endorse retrospective power calculations to aid interpretation of statistically non-significant trial results (see for example Hoenig and Heisey 20 ).
In common with many statistical methods, there is an underlying assumption of normality when using generalised linear models. This will hold for most adequately powered, phase III trials where analysis is completed on a scale for which the sampling distributions of estimated coefficients can be assumed normal (e.g. logistic, log). For small trials, or for estimands with very skewed distributions, asymptotic approximations may not hold and analyses should be checked using simulations.
In this paper, we provided expressions for the case where patients were randomised 1:1 to the experimental and control arms, although extension to other allocation ratios is straightforward. It would also be relatively straightforward to extend the methods to biomarkers with more than two levels, although the number of patients at each level is likely to be small in this case, resulting in low conditional power for all proposed rules. An exception might be for biomarkers with ordered levels, in which case the subgroup effect and interaction with treatment could be linear terms in the analysis (equation (1)).
For time-to-event outcomes, power of the study depends directly on the number of events occurring rather than on the number of patients, so that power would also depend on recruitment and censoring patterns. Methods would need to be extended to accommodate these features. Further, we have not embedded these results in more formal decision analytic methods, and this would require further specification of costs, harms (side effects) and utilities (benefits) and would depend on the perspective of the investigator (sponsor or health provider).
In summary, in situations where additional conditions are required for approval of a new treatment in a lower response subgroup, easily applied rules based on minimum effect sizes and relaxed interaction tests are available. These depend on trial design characteristics, particularly the proportion of patients sampled from the two subgroups and must be pre-specified and documented in the Statistical Analysis Plan.
Supplemental Material
sj-zip-1-smm-10.1177_09622802211017574 - Supplemental material for Frequentist rules for regulatory approval of subgroups in phase III trials: A fresh look at an old problem
Supplemental material, sj-zip-1-smm-10.1177_09622802211017574 for Frequentist rules for regulatory approval of subgroups in phase III trials: A fresh look at an old problem by K Edgar, D Jackson, K Rhodes, T Duffy, C-F Burman and LD Sharples in Statistical Methods in Medical Research
Footnotes
Acknowledgements
The project was stimulated by discussions with Sue-Jane Wang of the Food and Drug Administration and benefitted from discussions with David Wright of Astra Zeneca. The views and work contained within the paper are those of the authors. Software is available from the corresponding author upon request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: KE was supported by a UK National Institute for Health Research Methodology Fellowship.
Appendix 1. Conditional power calculations
The following shows how to calculate the conditional power for each rule, given pre-specified parameters
Given pre-specified treatment effects
We have that the conditional power for each rule is
The denominator does not depend on the form of any proposed rule and is given by
Note that the right-hand side is a function of three location parameters
For rule 1, the numerator of the conditional power is given by the expression
We make the transformation
Because ZF and
The covariance of X and Y is
Therefore, the joint distribution of X and Y is
The numerator is
The numerator for Rule 2 is given by
X is the same as for Rule 2, so that the expectation and variance of X are again
The expectation and variance of Y are
The covariance of X and Y is
The joint distribution of X and Y for Rule 2 is
Again, the numerator for conditional power is
For this rule the numerator is
We make the transformations,
Again the expectation and variance of X are
The expectation and variance of Y are
The covariance of X and Y is given by
Again, the numerator for conditional power is
To explore when conditional and unconditional power are the same, we identify conditions when
For Rule 3, conditional power is defined as
Recall that the covariance of X and Y is
If there is no correlation between the two subgroup treatment estimates ρ = 0, then this will become
Recall that for the normal distribution case with n patients allocated to each treatment arm and common sampling variance
For binary responses and usng a logit link,
In general, this will not hold. A similar situation applies for count data.
In summary, if the two subgroup estimates are correlated (
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.