
Abstract
In this paper, we develop a general Bayesian hierarchical model for bridging across patient subgroups in phase I oncology trials, for which preliminary information about the dose–toxicity relationship can be drawn from animal studies. Parameters that re-scale the doses to adjust for intrinsic differences in toxicity, either between animals and humans or between human subgroups, are introduced to each dose–toxicity model. Appropriate priors are specified for these scaling parameters, which capture the magnitude of uncertainty surrounding the animal-to-human translation and bridging assumption. After mapping data onto a common, ‘average’ human dosing scale, human dose–toxicity parameters are assumed to be exchangeable either with the standardised, animal study-specific parameters, or between themselves across human subgroups. Random-effects distributions are distinguished by different covariance matrices that reflect the between-study heterogeneity in animals and humans. Possibility of non-exchangeability is allowed to avoid inferences for extreme subgroups being overly influenced by their complementary data. We illustrate the proposed approach with hypothetical examples, and use simulation to compare the operating characteristics of trials analysed using our Bayesian model with several alternatives. Numerical results show that the proposed approach yields robust inferences, even when data from multiple sources are inconsistent and/or the bridging assumptions are incorrect.
1 Introduction
Bridging strategies are increasingly being used in the paradigm of global drug development1–4 to minimise duplication of clinical research without disregarding heterogeneity between patient groups. Bridging studies may be conducted in a new geographic region to evaluate whether a medicine’s performance (typically efficacy) is consistent with its performance in other parts of the world where it has been approved based on a complete development program. The International Conference on Harmonisation (ICH) E5 Guideline5,6 discusses whether and when trial data generated in an ‘original’ region can be leveraged to support the evaluation of drug activities in a new region where a sponsor is seeking registration. The degree of borrowing, ranging from none to full, is a matter of negotiation between the sponsor and the local health authority. By avoiding the unnecessary replication of evidence, bridging strategies can mitigate the drug lag problem7–9 and expedite patient access to new medicines.
Over the past few decades, the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan has promoted synchronisation of clinical drug development in Japan and other countries. 10 The agency encourages domestic sponsors to participate in global phase I dose-finding studies in oncology, which has led to a number of early phase bridging studies. It was further contended that phase I trials in Japan could be carried out in similar times as those in the west, based on the finding of small between-region heterogeneity in the toxicity profile of single agents, as 54 phase I oncology trials conducted at the National Cancer Center Hospital in Japan between 1995 and 2012 had been reviewed.11 In this paper, we will focus on the design and analysis of phase I bridging studies, which aim to support estimation of the maximum tolerated dose (MTD) in a new geographic region or patient subgroup of a previously studied disease indication. We want to leverage the dose–toxicity data available in relevant, already studied populations, without neglecting possible heterogeneity stemming from intrinsic factors such as a patient’s genetic make-up, and/or extrinsic factors such as diagnostic criteria and environmental exposures. Wider application of the proposed Bayesian model can also include, for example, phase I trials evaluating the toxicity profile of a given treatment for multiple disease subtypes.
Several model-based designs have been proposed for phase I clinical trials to account for potentially different safety profiles of a new medicine in various patient subgroups. Liu et al. 12 develop a bridging continual reassessment method (CRM) procedure that uses the dose–toxicity data from a completed historical trial to generate multiple sets of ‘skeleton’ probabilities for a new trial in a different geographic region, with the most plausible set of skeleton probabilities weighted favourably through the Bayesian model averaging. 13 Takeda and Morita 14 present a Bayesian dose-escalation procedure which dynamically leverages information from a historical study. Specifically, before the new trial begins, historical trial data are used to formulate a weakly informative prior for the parameter of a dose–toxicity model employed by the CRM for dose recommendations; so-called weakly informative because the prior effective sample size 15 is considerably smaller than the anticipated sample size of the new trial. Historical and new trial data are then linked through a ‘historical-to-current’ parameter, which reflects the degree of agreement between the studies.
Alternatively, relevant ‘complementary-data’ (or co-data for short) 16 can be leveraged from phase I clinical trials run concurrently with the trial of interest, or from commensurate patient subgroups enrolled in the same trial. O’Quigley et al. 17 propose a two-sample CRM to draw inferences about the MTD appropriate for each of the two non-overlapping subgroups of patients. The dose–toxicity curves are modelled through a pair of parameters, one of which represents information common to both subgroups and the other, as a ‘shift parameter’, for heterogeneity of the second subgroup in relation to the first. O’Quigley and Iasonos 18 discuss theoretical properties of this bridging model when the shift parameter is discrete, allowing for the recommended dose in the second subgroup to be one or two dose levels away from the estimate in the first. Wages et al. 19 extend this CRM-type shift model to account for uncertainty about the true shifts and design a phase I/II trial of stereotactic body radiation therapy, where the dose–response relationship may present as non-monotonic.
To date, designs for phase I bridging studies have focused on co-data from trials conducted under similar circumstances, for example, studies evaluating a different yet relevant patient subgroup. However, preliminary data from animal toxicology studies will also be available, as is required by regulatory authorities. 20 It is appealing to use the animal and external human trial data, in addition to any human trial data from relevant patient subgroups, so that dose recommendations at early stages of the phase I trial can be informed by all relevant evidence.21,22 The challenge is to properly link the dose–toxicity models for different animal species and human subgroups. In situations of strong differences between toxicity profiles, the co-data should be quickly discounted from the analysis of the new phase I clinical trial.
Zheng et al. 21 propose a robust Bayesian hierarchical model to leverage data from multiple animal species in a phase I oncology trial which will be performed in a homogeneous patient group, to support the interim and final dosing recommendations. In this paper, we extend their approach to accommodate the case that the study population is made of heterogeneous patient subgroups. The robust extention proposed in this paper can therefore augment a phase I bridging trial with co-data, which may comprise (i) data from completed preclinical animal studies and/or (ii) concurrent external data from either completed or ongoing trials conducted in related patient subgroups (e.g. patients from other geographic regions). When the intrinsic and extrinsic factors arising from ethnicity would result in heterogeneous dose–toxicity relationships, our model will estimate the subgroup-specific MTDs.
The remainder of this paper is structured as follows. In Section 2, we develop a robust Bayesian hierarchical random-effects model leveraging data from both animal studies and related human subgroups, to support the analysis of a new phase I oncology trial. In Section 3, we illustrate the use of the proposed methodology for improved decision making with various hypothetical data examples. In Section 4, we perform a simulation study to compare the operating characteristics of dose-escalation trials driven by the proposed model with several alternatives. Finally, we draw conclusions and look towards future research in Section 5.
2 Bayesian hierarchical model for animal data and heterogeneous human data
In this section, we generalise the Bayesian model of Zheng et al. 21 to leverage available animal data and dose–toxicity data from different human subgroups into new phase I clinical studies.
Suppose that at the time of planning a phase I clinical trial, M preclinical studies have been performed in K animal species, labelled
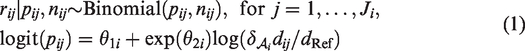
Random-effects distributions are stipulated on the second level of the hierarchical model to enable information sharing between animal studies of the same species

with
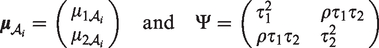

with
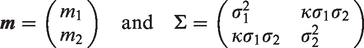
This ‘supra-species’ random-effects distribution accounts for the differences between toxicity parameters in different species which are not addressed by the translation parameters
We now focus on modelling the human toxicity data that will be collected from different human subgroups. Suppose there are a total of L predefined, non-overlapping human subgroups and one trial only is performed in each subgroup. To distinguish from the notation used for animal studies, we let
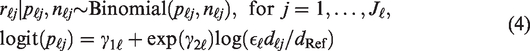
Here, instead of the log-normal priors which we specify for the animal-to-human translation parameters, we consider priors with a mode of 1 for
For simplicity, we assumed that one trial only had been undertaken per human subgroup. Following Neuenschwander et al.,
16
our model accommodates two exchangeability scenarios, along with one non-exchangeability scenario, for For
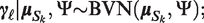
This represents exchangeability between With prior probability

where
With prior probability
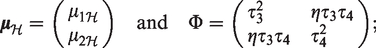
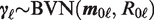
so that
The prior probabilities
The second ‘human only’ exchangeability distribution in our model has its own covariance matrix
We visualise the core of the proposed hierarchical model with a diagram in Figure 1, where
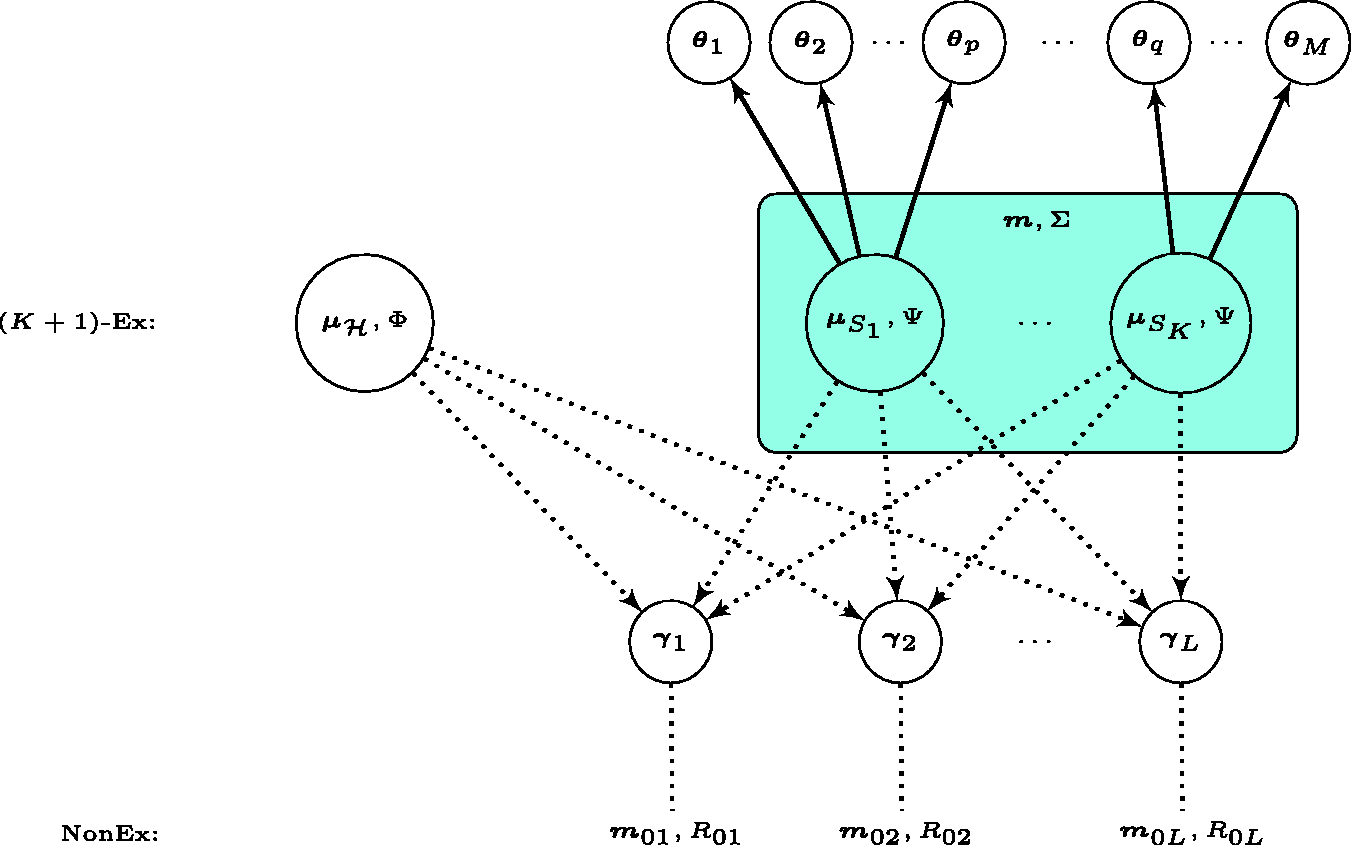
Diagram for the core of the proposed Bayesian hierarchical model. The solid (dotted) arrow suggests where a full (partial) exchangeability assumption holds. The possibility of non-exchangeability is enabled per human dose-toxicity parameter vector
To complete our Bayesian model, we now specify priors for other parameters. Weakly informative priors are placed on the hyperparameters of the random effects distributions in Models (3) and (5). The weakly informative priors used in subsequent sections are chosen so that each human toxicity risk
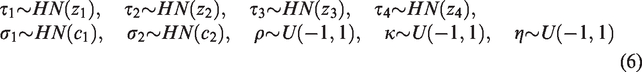
3 Illustrative example
In this section, we apply the robust Bayesian hierarchical model proposed in Section 2 to a hypothetical example informed by a real trial which aimed to characterise the toxicity profile of GSK3050002,
27
an antibody for treating patients with psoriatic arthritis. The original trial enrolled a total of 49 human subjects exclusively in the United Kingdom. For illustration, we assume that two hypothetical phase I trials (labelled
3.1 Hypothetical preclinical data and predictive priors for human DLT risks
According to the protocol of GSK3050002,
28
preclinical toxicity studies were performed in monkeys and rats. Moreover, monkeys were thought to be the most relevant animal species for predicting toxicity in humans. In the two real monkey studies, doses 1, 10, 30, 100 mg/kg were tested on 4–12 monkeys per dose group. From the trial protocol, it was not possible to identify what dose levels were used in rats, nor the exact number of rats treated, nor the number of toxicities observed. We therefore simulate plausible animal datasets based on the limited information available, and use these simulated data to obtain predictive priors for the human DLT risk at doses contained in the set
Throughout, we set
For a robust inference under scenarios of data inconsistency, independent non-exchangeability distributions BVN
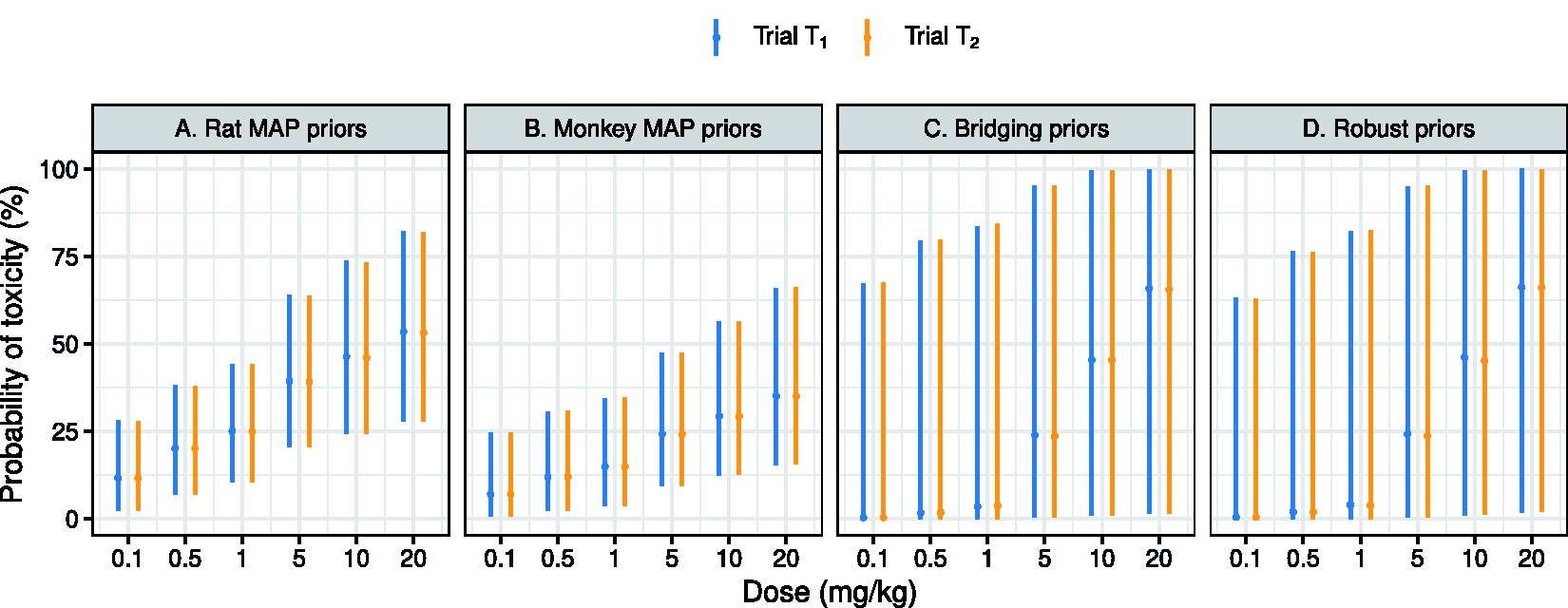
Summaries about the predictive priors for human toxicity, when using animal data from a single species (Panels A and B) or no animal data at all yet with a bridging assumption (Panel C) or without (Panel D). Medians together with 95% credible intervals of the marginal predictive priors are plotted.
As we can see, the rat and monkey data predict 1 mg/kg and 5 mg/kg as doses highly likely to result in a human DLT risk close to 25% in a human trial. After translation of the animal doses, rat data are mainly projected on the low doses of
We obtain MAP priors for the DLT risk in humans, by allocating prior weights to different animal species on the basis of their a priori predictability of the human toxicity. For trial
We characterise the predictive prior per dose
It will be helpful to assess the effective sample size (ESS)
15
of the predictive priors for each
3.2 Design and conduct of the phase I trials in different patient subgroups
Suppose that the phase I trials
Recall that we have estimated dose 0.1 mg/kg as a suitable starting dose for patients in cohort
Phase I trials will be terminated either after completion of treatment for all 24 patients, or for safety if for any dose (including the lowest dose) the posterior risk of overdosing is too high. When the complete data from trial
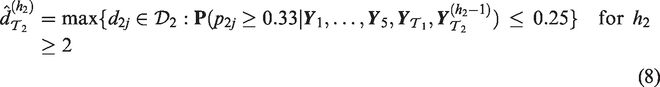
Figure 3 shows two simulated realisations of trials
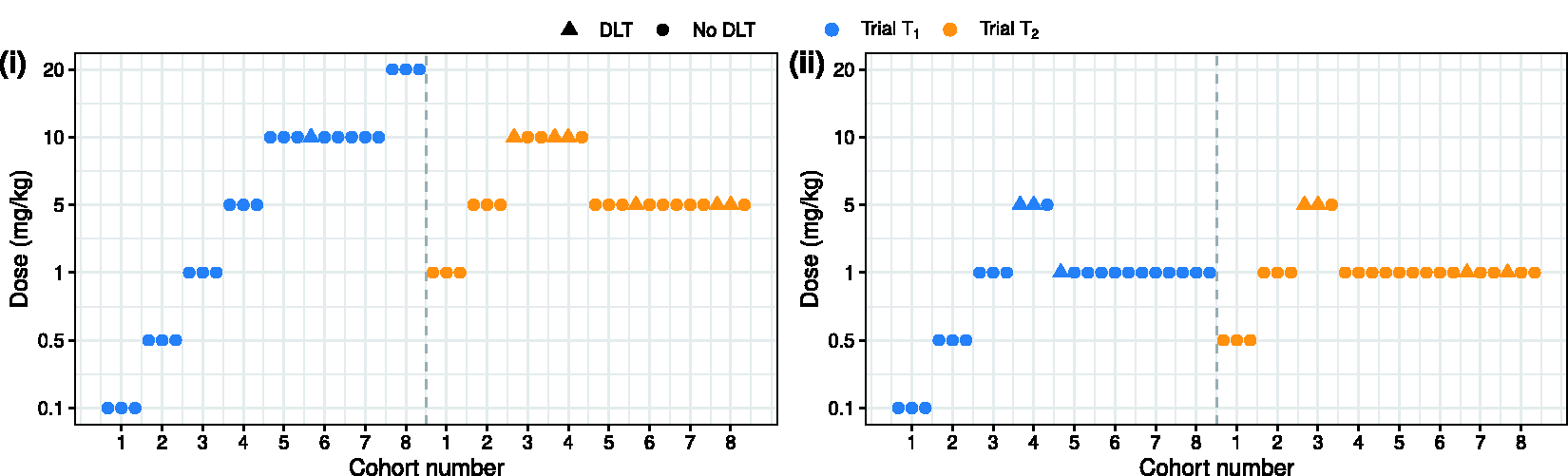
Trial trajectory of hypothetical phase I trials performed in two geographic regions, in which trial data were simulated from (i) a divergent scenario and (ii) a consistent scenario, respectively.
Operating characteristics of the adaptive phase I dose-escalation trials in regions
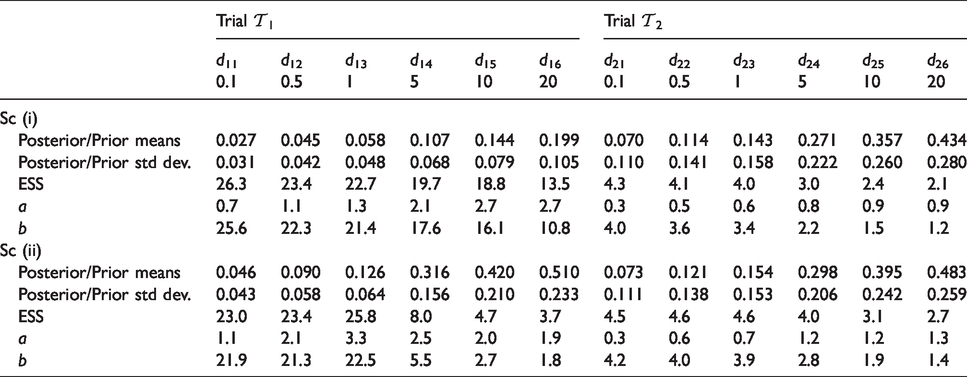
On the completion of trial
Effective sample sizes of the marginal predictive posteriors (priors) for the DLT risk per dose, on the completion of trial
4 Simulation study
In this section, we compare the operating characteristics of phase I dose-escalation trials, conducted using the proposed Bayesian hierarchical model or an alternative. The analysis models we consider are as follows:
Model A is the proposed Bayesian model leveraging co-data from multiple sources; Model B discards animal data and assumes human parameter vectors Model C analyses trials Model D leverages animal data for trials Model E analyses trial
The prior specifications for Model A remain unchanged from Section 3.1. All the simulated
Each simulated phase I trial is performed in an adaptive manner: interim dose recommendations are made according to criteria (7) and (8) for trials 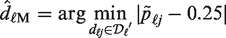
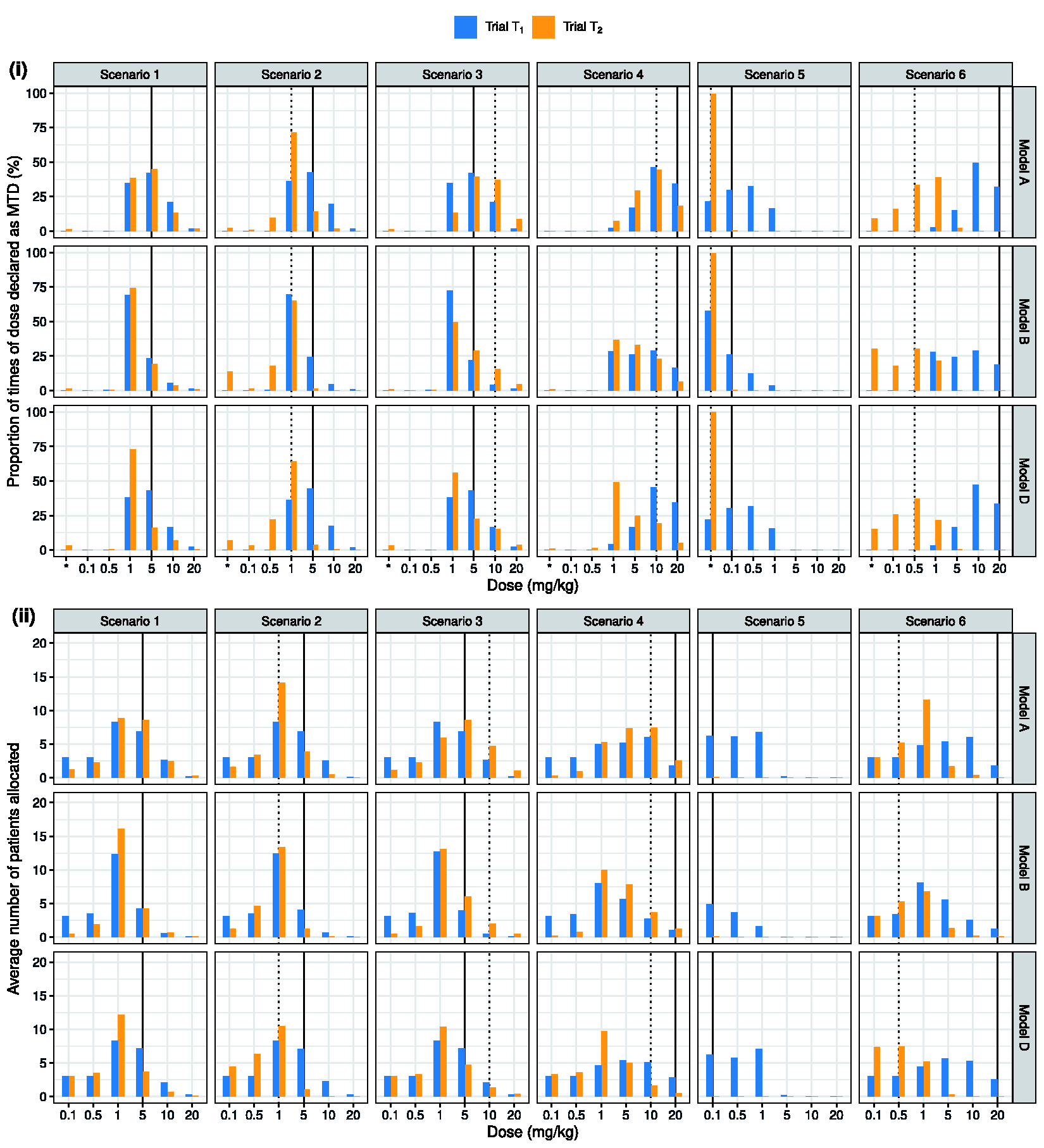
Simulation scenarios for the true probability of toxicity in humans for the phase I trials
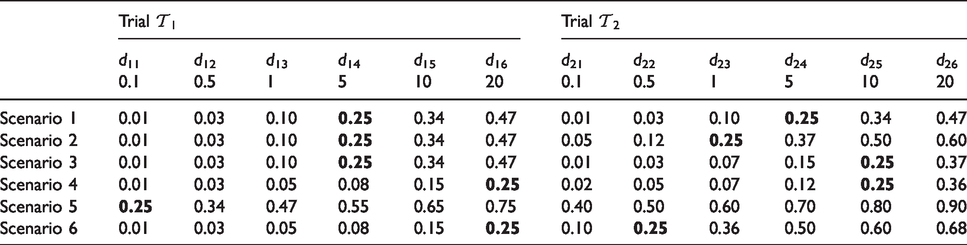
For each toxicity scenario, we simulated 1000 pairs of adaptive phase I dose-escalation trials in regions
Complete results from the simulation study can be found in Table S2 of the Supplementary Materials, and comparisons between Models A, C and E are presented in Figure S3. Here, we focus on comparing the operating characteristics of trials driven by Models A, B and D shown in Figure 4. We see that Model A outperforms the alternative analysis models across nearly all the simulation scenarios. In scenarios 1 and 2, where animal data are highly predictive of human DLT risks, Models A and D which leverage animal data lead to a higher percentage of correct selection (PCS) and a higher proportion of patients allocated to tolerable doses with DLT risks in the range [0.16, 0.33) than Model B. Comparing Models A and D, allowing for information sharing across patient subgroups leads to an increase in the PCS in trial
In scenario 5, all the Bayesian analysis models (A–E) limit the exposure of patients to overly toxic doses, say, doses with a DLT risk exceeding 50%. Due to the use of animal data, Models A and D tend to treat more patients than Model B with doses 0.5 and 1 mg/kg, which have human DLT risks exceeding 33%. However, the average number of patients experiencing a DLT is not substantially higher than the number under Model B. Scenario 6 represents the case where the bridging assumption is incorrect. Comparing trial operating characteristics under Model A with those under Models C and D, we find trials driven by Model A allocate 6–7 more patients to dose 1 mg/kg in trial
In scenarios 1–3, Model B assigned more patients in trial
Referring to the Supplementary Materials, we can draw comparisons between the operating characteristics of dose-escalation procedures driven by Model A versus Models C and E. Models C and E can be regarded as extremes, with either permit no borrowing at all or complete pooling of human data across regions. The improved operating characteristics when comparing Models A and C should be interpreted as a mixture of the benefit from using both animal data and an appropriate bridging strategy. We have also compared Models A–E with respect to the posterior median estimates of the human DLT risks in each region, dose–toxicity relationship in each human subgroup. Figure S4 of the Supplementary Materials show that Model A outperforms the others, providing very satisfactory characterisation of the association on the termination of a phase I clinical trial. We additionally ran simulations for a robust version of Model B with
We introduced bridging parameters
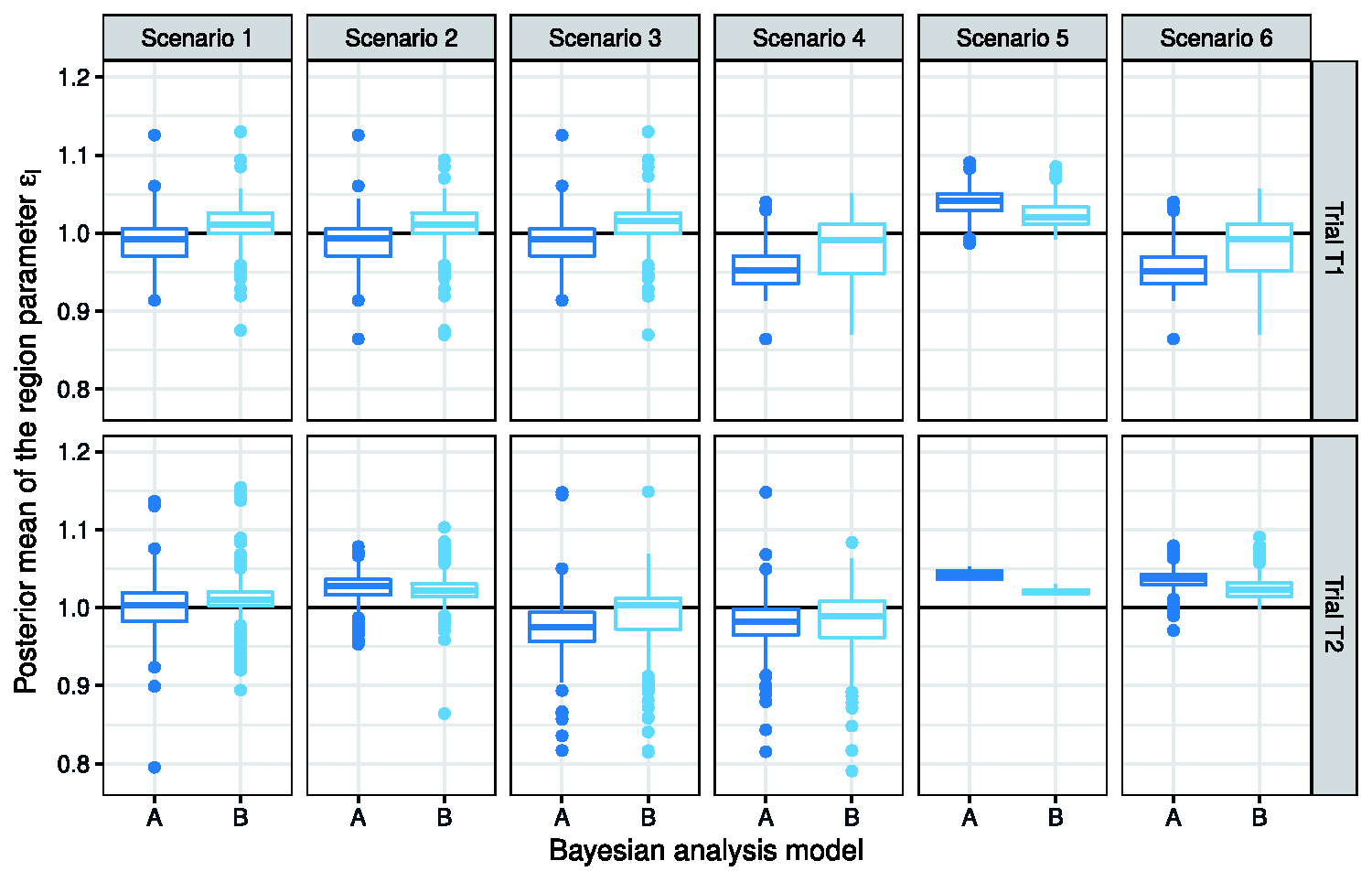
Boxplots that depict the posterior means of the region parameter
Additional simulations were performed to support future application of the proposed Bayesian hierarchical model. In particular, we evaluated the impact of prior probabilities of exchangeability and non-exchangeability, i.e.
5 Discussion
Bridging studies have received considerable interest, 32 as fewer resources may be needed to demonstrate drug behaviours by using relevant data from other subgroups, compared with the approach of establishing an independent, complete package of clinical drug development. Statistical methodology to extrapolate across geographic regions has been proposed mainly in the context of phase II and phase III clinical trials.33–36 Much less has been written on phase I clinical trials, where different metrics are used to evaluate trial efficiency and estimation accuracy.
In this paper, we seek to improve decision making in a phase I bridging study, by leveraging not only the trial data on an original subgroup/region for drug registration, but also preclinical animal data. The novelty of the proposed methodology is relating to sensible constellations of parameter vectors. Technically speaking, the hierarchy of the proposed model is constructed by placing the human dose–toxicity parameter vectors
In our simulation study, we use the prior probabilities
We note that the proposed methodology has wider applications. For example, there may be a need to design phase I dose-escalation trials in subgroups defined by clinical or genetical characteristics which could potentially modify the therapeutic effect of the drug.
37
Based on our Bayesian model, information from patient subgroups with similar safety profiles can be leveraged. There is no restriction on the number of studies that will be run in the new patient subgroups, nor on the number of subgroups to provide the co-data. When a large number of subgroups are involved, estimate of parameters that represent the between-trial heterogeneity (specifically,
This paper has focused on the design of bridging studies to estimate the MTD in a new geographic region or human subgroup of a previously studied disease indication, although the approach can also be used for other settings. Future work could consider extending the proposed hierarchical model to accommodate the case of bridging across subgroups in related disease indications, when distinct endpoints, different dosing schedules or formulations, etc. might be necessary. The research question is highly relevant within the paradigm for precision medicine. A new class of efficient approaches, known as basket trials, 38 have emerged, where the same treatment is tested in potentially heterogeneous patient subgroups (often defined by genetic characteristics). Robust hierarchical models have been considered for borrowing of information.39,40 The proposed methodology can potentially be used to analyse phase I oncology basket trials, where multiple cancer subtypes are studied under a master protocol. 41 It is conceptually similar to the proposal by Neuenschwander et al. 39 : prior probabilities of exchangeability and non-exchangeability are assigned independently to each vector of subgroup-specific dose–toxicity model parameters. Our Bayesian model allows co-data to contribute towards formulating the exchangeability distributions so as to discuss borrowing of information from specific sources. Independent non-exchangeability distributions ensure we obtain the robust estimates of the dose–toxicity model parameters underpinning extreme subgroups. Improving statistical inferences for extreme subgroups, which could be similar amongst themselves, is outside of the scope of the present research. This is an area for future research, with related investigation undertaken in the context of phase II basket trials to enable information sharing based on distributional discrepancy between model parameters for therapeutic effects. 42
Supplemental Material
sj-pdf-1-smm-10.1177_0962280220986580 - Supplemental material for Bridging across patient subgroups in phase I oncology trials that incorporate animal data
Supplemental material, sj-pdf-1-smm-10.1177_0962280220986580 for Bridging across patient subgroups in phase I oncology trials that incorporate animal data by Haiyan Zheng, Lisa V Hampson and Thomas Jaki in Statistical Methods in Medical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 633567. Dr Hampson’s contribution to this manuscript was supported by the UK Medical Research Council (grant MR/M013510/1). This report is independent research arising in part from Prof Jaki’s Senior Research Fellowship (NIHR-SRF-2015-08-001) supported by the National Institute for Health Research. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health and Social Care (DHSC). T Jaki also received funding from the UK Medical Research Council (MC__UU__0002/14).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.