
Abstract
Information on the age at onset distribution of the asymptomatic stage of a disease can be of paramount importance in early detection and timely management of that disease. However, accurately estimating this distribution is challenging, because the asymptomatic stage is difficult to recognize for the patient and is often detected as an incidental finding or in case of recommended screening; the age at onset is often interval-censored. In this paper, we propose a method for the estimation of the age at onset distribution of the asymptomatic stage of a genetic disease based on ascertained pedigree data that take into account the way the data are ascertained to overcome selection bias. Simulation studies show that the estimates seem to be asymptotically unbiased. Our work is motivated by the analysis of data on facioscapulohumeral muscular dystrophy, a genetic muscle disorder. In our application, carriers of the genetic causal variant are identified through genetic screening of the relatives of symptomatic carriers and their disease status is determined by a medical examination. The estimates reveal an early age at onset of the asymptomatic stage of facioscapulohumeral muscular dystrophy.
1. Introduction
A reliable and accurate estimate of the age at onset distribution of a disease is of great importance for optimizing follow-up protocols of high-risk patients, aiming at early detection of the disease and timely start of treatment. For most diseases treatment is more effective if it is started in an early stage than at a later moment when the disease has progressed.
Diseases usually progress in stages. The early stage is typically asymptomatic. In this phase of the disease the patient is not aware of having the disease, because no symptoms with which the disease is usually associated are experienced. Diagnosis is most often in the symptomatic stage when symptoms appear. Although the disease is not apparent during the asymptomatic stage, some pathological changes may be detectable with a medical test. For common diseases like breast and colon cancer, population screening programs have been set up in many countries to identify the disease process during this asymptomatic phase so that intervention can be started at an early stage in the disease process. However, population screening is only offered for a limited scope of diseases, because of cost-effectiveness and possible profit for the patient. Small-scale screening is performed in high-risk sub-populations, typically characterized for certain rare genetic variants.1,2
Since genetic traits aggregate within families, these high-risk sub-populations are often identified via family members carrying the genetic variant and being affected by the disease. Specifically, families who satisfy certain ascertainment criteria (i.e. “at least one affected carrier”) are selected and all family members (up to a certain degree) are invited for a genetic test. Individuals who carry the genetic variant of interest may be then (regularly) screened for the presence of the disease in the asymptomatic stage. The screening scheme and especially the age at which screening starts should depend on the risk the carrier will become asymptomatic. To determine this risk, an estimate of the age at onset distribution for the asymptomatic stage is needed.
Estimation of this age at onset distribution for the asymptomatic stage is challenging, because it should rely on data of selected families (based on the presence of the disease). Moreover, the exact age at onset is never observed for any of the individuals in the data-set. Instead, at every moment of screening it is observed whether an individual has the disease in asymptomatic stage or not; age at onset is interval-censored by the ages at time of screening. Sometimes, if the disease is not lethal or no treatment is available, individuals are screened only once. Then, only the age at time of screening is observed, as well as whether or not the individual is asymptomatic before examination took place. This type of censoring is referred to as interval-censored type I or current status. 3
With interval-censored data, the exact age at onset is never observed for any individual; therefore, this kind of censoring differs greatly from right censoring. If the data are obtained from a population of independent, non-selected individuals, for instance with population screening, the distribution of the age at onset of the asymptomatic disease can be estimated with the non-parametric maximum likelihood estimator (NPMLE) (see, e.g. Zhang and Sun, 3 Groeneboom and Wellner, 4 Jewell and van der Laan, 5 and Witte et al. 6 ). In case of selected families, one often tries to correct for ascertainment bias by leaving out the index patients and assuming independence between the relatives. Under this independence assumption, the NPMLE could be used, but the estimator is still biased because of the suboptimal ascertainment correction (leaving out the index): it does not take into account the fact that ascertainment is based on the phenotype of all individuals in the pedigree and not on one particular person. The bias is especially large in data-sets with small pedigrees. 7 Moreover, leaving out the data of the index patients from the analysis, means that valuable information is discarded, what is especially unfortunate in rare diseases for which data-sets are often small. A more sophisticated analysis has to be performed to properly correct for ascertainment and to use the data optimally. This could be done by considering an adjusted likelihood which conditions on the ascertainment event, for instance by maximizing the retrospective likelihood, the prospective likelihood, or a joint conditional likelihood (e.g. Carayol and Bonaiti-Pellie 8 and Kraft and Thomas 9 ).
In this paper, we propose a maximum likelihood-based method, adjusted for the ascertainment, to estimate the distribution of the age at onset of the asymptomatic stage of a disease. The likelihood function has a complex form as it takes into account all the available information: the interval-censored age at onset of the asymptomatic stage, the (right-censored) age at onset of the symptomatic stage, the ascertainment criteria, and also family characteristics that may affect the age at onset distributions. Simulation studies are performed to study the performance of the proposed estimator.
For many rare disease susceptible genetic variants, the number of detected pedigrees with this variant is small, like for the disorder that motivated this research. This complicates estimation of the age at onset distributions and hence the proposed statistical model cannot be too large (too many unknown parameters) to overcome overfitting of the data. A balance has to be found between the complexity of the model and the information in the data. The proposed model can also be applied if the sample size is small.
This work is motivated by a study on facioscapulohumeral muscular dystrophy (FSHD), a genetic muscle disorder. In order to learn more about the progression and causes of the disease, a cross-sectional observational study of families with at least two symptomatic family members was performed. All family members were offered a genetic test and a physical examination to determine if they had already entered the asymptomatic stage of the disease. No extra data were collected after the moment of examination. The age at onset for the asymptomatic stage is hence type I interval-censored. The date of examination was the same for all the participants in the study (cross-sectional).
The rest of the paper is organized as follows. In the second section, we introduce notation and establish a general framework for the problem. Ascertained-corrected conditional likelihood estimation is proposed in the third section. A simulation study is presented in the fourth section, while in the fifth section the methods are applied to a data-set of familial FSHD in the Netherlands. Main conclusions and a final discussion follow in the final section.
2. Data, notation, and assumptions
In case of genetic diseases, carriers of the disease susceptible variant are often identified via already detected carriers; relatives of affected carriers are invited for genetic testing. For data analysis and estimation of the age at onset distribution of the disease, usually only data of the proven carriers from ascertained families are included in the data-set (and the non-carriers are left out). Below, notation for carriers of the causal variant is introduced.
Let U and T be the ages at onset of the asymptomatic and symptomatic stage, with
The three possible configurations of observations are C < U < T,
We assume that all individuals with a positive genetic test for the variant of interest are physically examined and included in the data-set. For a single carrier, the triple
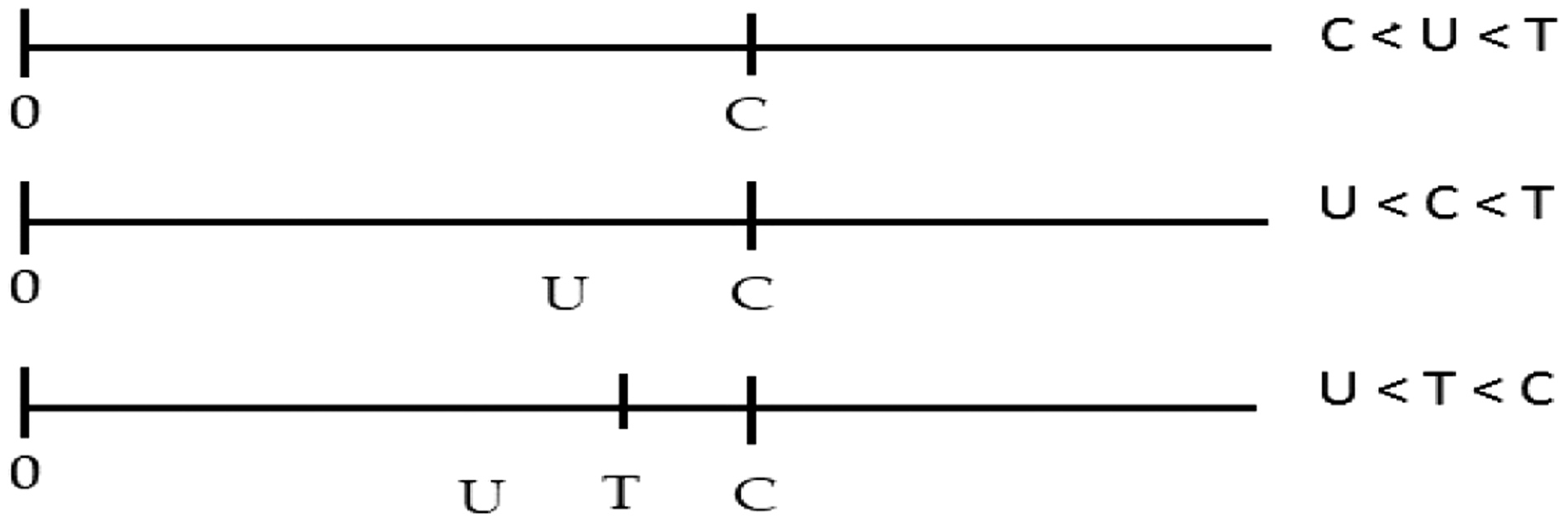
Presentation of the observations in the three configurations. There are no observations after C and the exact moment U took place is not indicated, because this is unknown.
We assume that U and T follow (parametric) distributions,
Definition of variables and distributions.

In the paper it is assumed that, conditional on the covariates and the genetic status of the individuals, the ages at onset of the asymptomatic and the symptomatic stages are independent between individuals.
For tested non-carriers, the ages at time of genetic testing are known (from the records). These data can be used for estimating the distribution G. This is under the assumption that the distributions of the age at testing are equal for carriers and non-carriers. This is a reasonable assumption, because carriers and non-carriers do not know their carrier status when they decide to be genetically tested or not.
3. Ascertained-corrected conditional likelihood estimation
Our main goal is to obtain an accurate estimator of the distribution of the age at onset of the asymptomatic stage for carriers of the variant,
In the derivation of the likelihood functions, we assume that, conditional on the covariates, the observations of all carriers are independent, notwithstanding the pedigrees they belong to. In the notation, no discrimination with respect to the pedigrees is necessary and all carriers are identified with a single and unique index i: the variables
In Appendix 1, a derivation is given of the ascertainment-corrected prospective likelihood functions Lsymp and Lfull. The likelihood function based on the symptomatic data 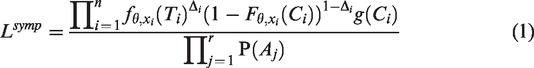
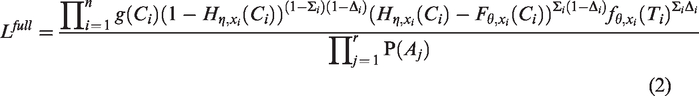
Since the pedigrees are not randomly sampled from the population, the likelihood functions are conditional on the ascertainment events 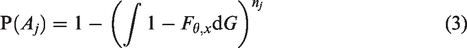
In Section 5, we give an explicit expression of
3.1. Estimation of parameters
From a theoretical perspective the full likelihood, Lfull, could be maximized with respect to all unknown parameters to obtain their maximum likelihood estimates. However, in practice we noticed that optimization algorithms often do not converge to the global maximum, probably because the parameter space is too big and the algorithm stops at a local and not the global maximum. Therefore, we propose to perform the estimation of the unknown parameters in the model in two steps. Simulation studies in Section 4 show good performance of this two-step procedure. The two steps are given by:
Distribution G and parameter θ are estimated based on the likelihood function Lsymp in equation (1). More details are given below. After inserting the estimates
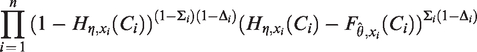
with respect to η.
In the first step of the estimation algorithm, G and θ are estimated without using data on the asymptomatic status (Σ). In principle, this does not affect the unbiasedness of the estimators, but because not all information is used standard errors might slightly increase. If G is assumed to follow a parametric distribution, G and θ can be estimated by maximizing the likelihood function Lsymp in equation (1). Alternatively, G could be estimated by the empirical distribution of the ages at time of examination of the relatives with a negative genetic test (non-carriers). This estimator is asymptotically unbiased. If no data of non-carriers are available, the ages at examination of the carriers could be used instead. This yields an estimator that is asymptotically slightly biased. An extensive discussion on this topic, including simulation studies, is given in Jonker et al. 7
Our two-step estimation procedure implies that two maximizations are required, but the dimension of the parameters space in each maximization problem is reduced. This lowers the computational complexity of the problem. Of course, the likelihood Lfull in equation (2) could also be maximized with respect to all parameters simultaneously, but, as mentioned before, the two-step procedure shows more stable results (this has been checked with simulations).
3.2. Variance estimation
Variance estimates of Randomly select a pedigree from the original data-set. Say pedigree j is selected. For each of the nj family members i of the selected pedigree j, draw To guarantee that the resulting bootstrap resample is similar to the original data-set in terms of family size and structure, the previous step is repeated until the simulated phenotypes of the carriers in the pedigree satisfy the ascertainment condition.
These three steps are repeated until the bootstrap resample contains the same number of pedigrees as the original data-set. For each bootstrap resample b, we apply the proposed estimation procedure described in Section 3.1. to obtain bootstrap estimates of the parameters of interest
By constructing the confidence intervals for the unknown parameters in this way, the inaccuracy of the estimators for G and θ is reflected in the width of the confidence interval for η.
4. Simulation study
We conduct a simulation study to illustrate the performance of the proposed estimation procedure. The setup and the results are described below.
4.1. Simulation setup
We assume that the variables for age at onset of the asymptomatic and symptomatic stages, U and T, follow gamma distributions. Further, inspired by our real data example, we assume that the variable for the age at time of examination, C, is independent of (U, T). We consider three basic scenarios which resemble relevant situations in practice. In scenario 1, we assume a situation in which the asymptomatic stage is short; the onset of the asymptomatic and symptomatic phases of the disease is close to each other. Namely, we assume that the expected age at onset of the asymptomatic stage, U, has mean
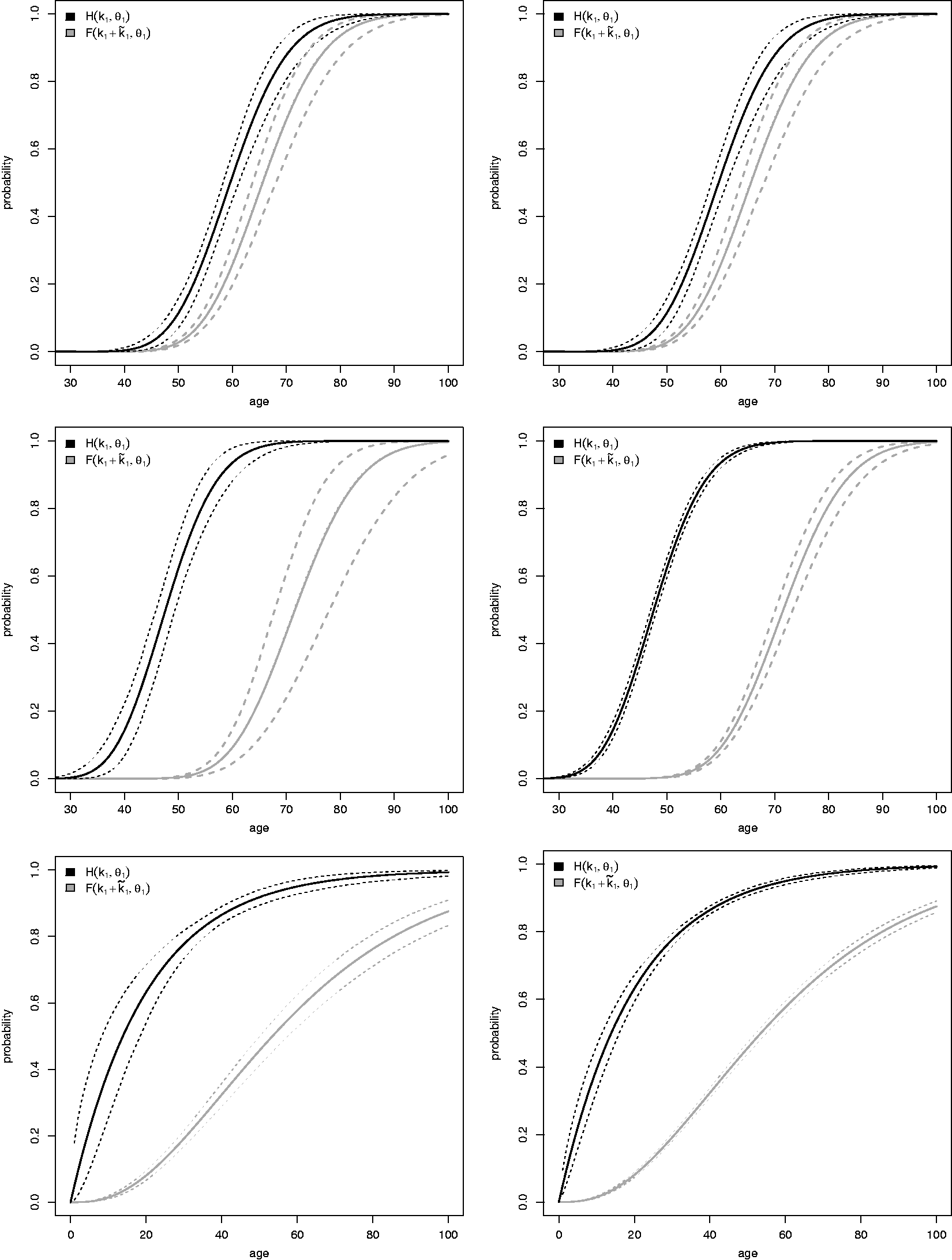
Results of the simulation study with m = 500 families. Top: scenario 1 (
We simulate M = 1000 Monte Carlo trials, each consisting of m families (m is taken equal to 100, 500, or 1000). Additionally, in scenario 3 we also considered m = 25 to mimic our real data setting with regard to the reduced number of observed families. m = 25 was not considered in scenarios 1 and 2 since it led to extremely low numbers of observed families so that inference was meaningless.
For each family j, Simulate family size nj. In order to check the impact of the family size on the performance of our method, two situations are considered: populations composed of “small” families (nj is sampled from For each family member For each family member i, For each family member i, For each family member i, If the ascertainment event Aj occurred, the nj carriers of family j are ascertained. In our simulation setting, families are selected if at least one symptomatic family member is identified (at least one family member with Independently of the simulations in the previous steps, a sample of 1000 observations from the uniform distribution at [20, 70] is simulated (the same distribution from which was simulated in step 5). The simulated values represent the ages at time of genetic testing of the individuals who got a negative test result. These data are used to estimate G (as explained in Section 3.1).
In the simulation study, families are ascertained if at least one family member is symptomatic at the time of examination, like in the example given earlier. As a result of the ascertainment, the effective sample that is used for estimation is smaller than m families; the effective sample size is denoted by r. To compute confidence intervals of the estimators, we set the number of bootstrap resamples equal to B = 500.
In practice, often some carriers in an ascertained pedigree do not participate in the study. It is likely that those carriers do not show symptoms of the disease. To check the impact of this form of informative missing data, we perform a second simulation study in which every non-symptomatic family member (Δ = 0) is excluded from the sample with a probability of either 0.20 or 0.50.
All computations are performed using the statistical software R (R Core Team, 2018). The function
4.2. Simulation results
The main results of the simulation study are shown in Figure 2 and in Table 2. In each graphic in Figure 2, the solid black and gray lines represent the true distributions of
reBias, standard deviation (SD), and coverage probabilities of the 95% confidence intervals (Cov) for the location and scale parameters of the age at onset gamma distributions of U (k1, θ1) and T (
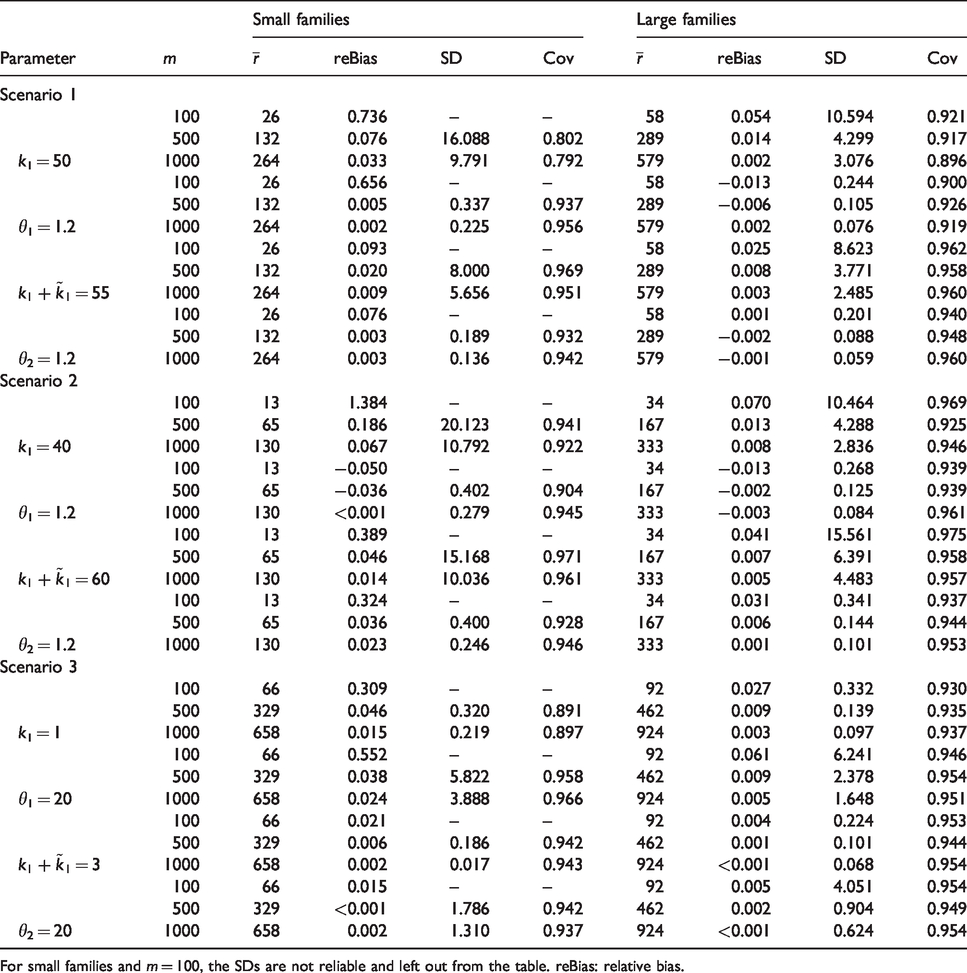
For small families and m = 100, the SDs are not reliable and left out from the table. reBias: relative bias.
From these graphics, the proposed estimators seem to be unbiased. The median estimated curves based on the median parameter estimate along the M = 1000 Monte Carlo trials cannot be distinguished from the theoretical distributions, and the bands formed by the 2.5 and 97.5% Monte Carlo percentiles nicely cover the theoretical curves in all the studied scenarios. In each Monte Carlo trial, data of m = 1000 families are simulated. Since not all of these m families satisfied the ascertainment criteria, the effective number of families included for analysis in each Monte Carlo trial, denoted as r, is considerably lower (see Table 2 for details). This might be the reason for the wide percentile band in the right upper graphic in Figure 2.
Table 2 complements Figure 2 and provides further results of the simulation study. For each of the studied scenarios, we provide results on mean estimated relative bias (reBias) (defined as the difference between the simulated mean and true parameter value divided by the true value), empirical standard deviation, and coverage probabilities across the 1000 Monte Carlo trials of the scale and shape parameters of the distribution of
Special mention deserves scenario 3, large families and m = 25 (corresponding to a mean number of ascertained families along the 1000 trials of
In the second simulation study, we evaluate the level of introduced bias in our estimates due to informative missing. For scenario 1 (
5. Motivating example
This work was motivated by a study on FSHD, a genetic muscle disorder. The severity of this disease is associated with a specific form of genetic lesion, the loss of repetitions of the D4Z4 unit. 10 Individuals without a loss of units (they have at least 10 units) are considered to be healthy and are not susceptible to develop the muscle disorder. It is expected that the age at onset of the asymptomatic and the symptomatic stage is also associated with the number of repetitions of this unit.
The data come from a cross-sectional study in which at a fixed and non-informative calendar time, all affected pedigrees in the Netherlands with at least one affected member among the first and second degree of the index patient (the first diagnosed patient in a family) were invited to participate in the study (see Wohlgemuth et al. 11 for details). So, the ascertained families have at least two affected individuals with both a loss of repetitions of the D4Z4 unit: the index patient and a relative. Data of 10 pedigrees consisting of in total 155 individuals are available. Of these 155 individuals, 69 present loss of repetitions of the D4Z4 unit at some degree (the so-called carriers) and 86 have no genetic alteration (the non-carriers). All individuals within a pedigree with a loss of repetitions have an equal number of repetitions. An overview of the carrier-data is given in Table 3.
Overview of data: For every pedigree, the number of units, carriers, and symptomatic and asymptomatic carriers are given.
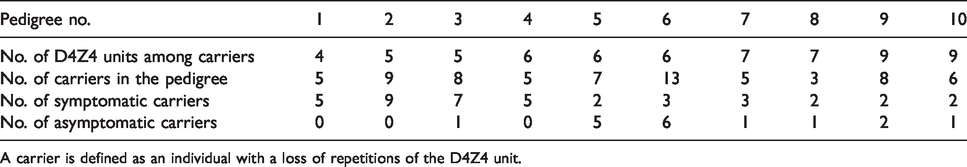
A carrier is defined as an individual with a loss of repetitions of the D4Z4 unit.
We consider two parametric models: the Weibull distribution for both
For nj the number of individuals in pedigree j in the data-set, the probability the ascertainment event occurs (at least two symptomatic patients at examination), equals 1 minus the probability that none or only one of the nj individuals is symptomatic
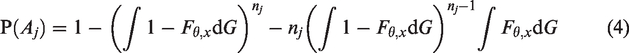
We estimate G by the empirical distribution function of the ages at time of examination C of the individuals with and without a loss of repetitions together. Since the number of observations is low, we chose to combine the data when estimating G. Next, we follow the estimation procedure as described in Section 3.
Based on the value of the likelihood (or AIC), the gamma model fits slightly better than the Weibull model. The actual estimates of
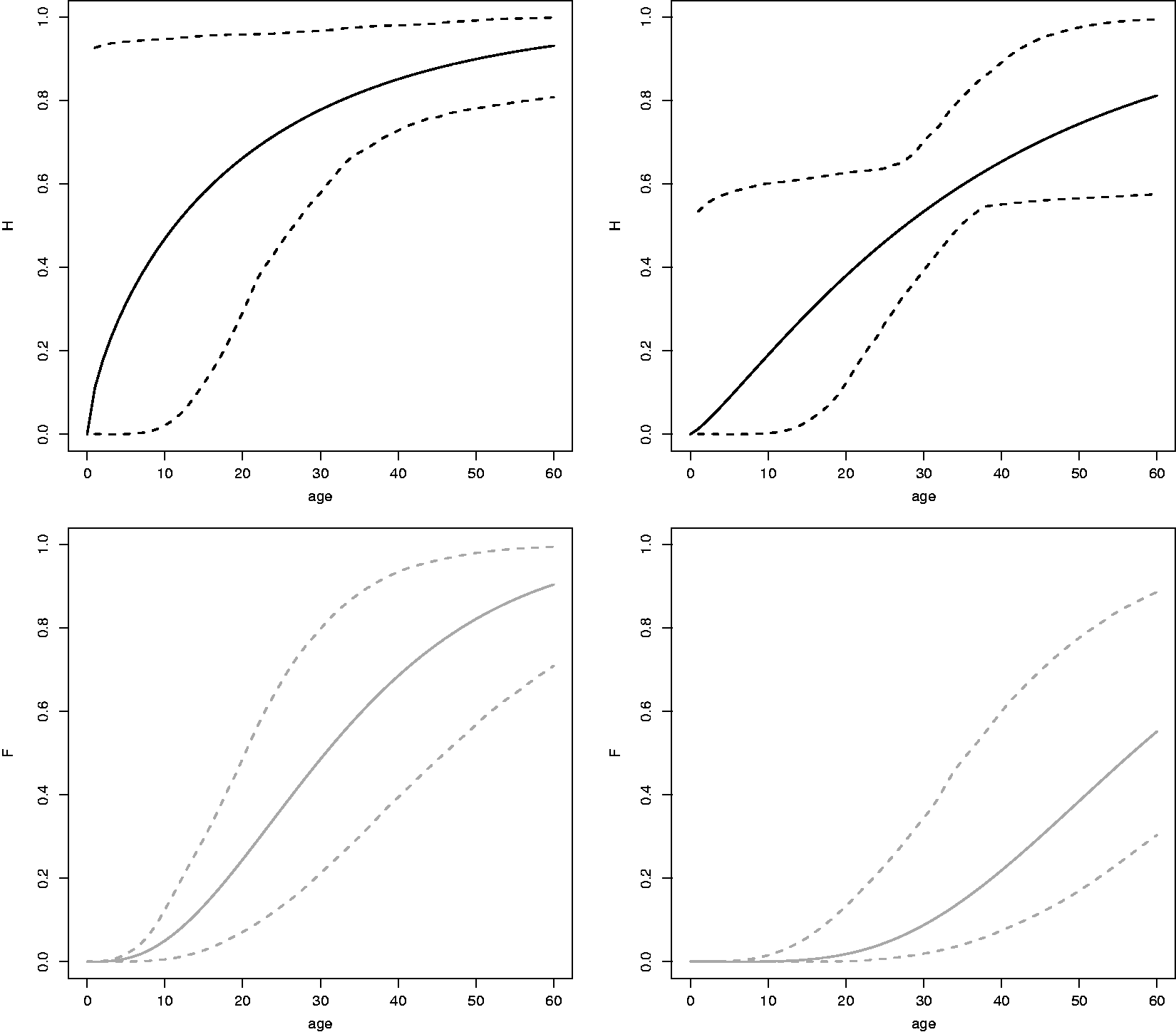
Solid lines: Estimates of
The estimates of the age at onset distribution of the asymptomatic and the symptomatic stage of FSHD show that these functions depend on the covariate repeat size and both increase until late adulthood. These estimates can be used in counseling and help in understanding progression of the disease over time. However, the number of individuals on which the estimates are based is low and should be interpreted with care.
R-code for maximizing the log-likelihood functions is provided in Appendix 4.
6. Discussion
In this paper, we have proposed a maximum likelihood-based method for estimating the age at onset distribution for the asymptomatic stage of a genetic disease using clinically ascertained pedigree data. Simulation studies showed that as long as the sample is not too small, our estimation method yields accurate results. Estimates of this distribution are of great importance for setting up follow-up programs in high-risk families, for instance families with a genetic variant associated with susceptibility of a disease.
The estimates of the age at onset distribution of the asymptomatic and symptomatic stage of FSHD, found in the application, can be used to learn more about the progression of the disease. Since there is no treatment available and the disease is not life threatening, one could argue whether screening is necessary, but eventually this decision will be made by the family and their medical doctor.
In this paper, we considered the situation of a cross-sectional study which took place at a randomly chosen moment (set by the researcher), without any follow-up afterwards. However, regular screening of patients with a high disposition of a slowly developing disease is quite common.1,2 To fit these kind of screening data, the expressions of the likelihood functions need to be adjusted, but the underlying principle of estimating the age at onset distributions for the asymptomatic and the symptomatic stage in two steps remains valid.
Measured family characteristics can be included in the model via covariates. To account for unmeasured family characteristics, a frailty term (random family effect) could be added to the model (see, e.g. Gong et al., 12 Hsu et al., 13 Hsu and Gorfine, 14 and Gorfine et al. 15 ). In a shared frailty model, every family has its own frailty that describes the susceptibility of the family members to develop the disease compared to the population of interest. This model could be further generalized to correlated frailty models in which every individual in the family has its own frailty term, but these terms are correlated within families (and are independent between families). This gives the model more flexibility. However, for fitting these models sufficient data must be available; the number of pedigrees and the number of individuals in the pedigrees must be sufficiently large. This is certainly not the case in our application, but including frailties in the model is an interesting topic for further research.
Since we are considering the distinct stages (healthy, asymptomatic, symptomatic), our data could be modeled with a multi-state model. 16 However, as far as we know, multi-state modeling with data that are ascertained based on the outcome is still an open problem.
In this paper, we have assumed that the medical tests at screening moments are fully sensitive and specific. In population screening programs like breast and colon cancer, the screening tests are often imperfect; the sensitivity and specificity of the test are below 100%. 6 To preclude unnecessary treatment, a screening test is followed by a confirmative test in case of a positive screening test. The confirmative test is assumed to be 100% sensitive and specific. In family studies, only high-risk individuals (carriers) are screened. For this purpose usually the most accurate medical test is applied and an assumption of full sensitivity and specificity is reasonable. Otherwise, the expression of the likelihood must be adapted, but the methodology will be the same.
To conclude, reliable estimates of the age at onset distribution of the asymptomatic stage are important for setting up personal follow-up screening in high-risk families. The methodology described in this paper is therefore of great relevance.
Footnotes
Acknowledgements
We like to thank M. Wohlgemuth and N. Voermans for collecting and making available the FSHD-data that were analyzed in Section 5. Further, we like to thank the reviewers for their helpful comments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.