
Abstract
The number of trials conducted and the number of patients per trial are typically small in paediatric clinical studies. This is due to ethical constraints and the complexity of the medical process for treating children. While incorporating prior knowledge from adults may be extremely valuable, this must be done carefully. In this paper, we propose a unified method for designing and analysing dose-finding trials in paediatrics, while bridging information from adults. The dose-range is calculated under three extrapolation options, linear, allometry and maturation adjustment, using adult pharmacokinetic data. To do this, it is assumed that target exposures are the same in both populations. The working model and prior distribution parameters of the dose–toxicity and dose–efficacy relationships are obtained using early-phase adult toxicity and efficacy data at several dose levels. Priors are integrated into the dose-finding process through Bayesian model selection or adaptive priors. This calibrates the model to adjust for misspecification, if the adult and pediatric data are very different. We performed a simulation study which indicates that incorporating prior adult information in this way may improve dose selection in children.
1 Introduction
Phase I dose-finding studies represent the first transition from laboratory work to a clinical setting and aim to obtain reliable information on the pharmacokinetics (PK), safety and tolerability of a drug. Typically, these trials are performed on healthy subjects, unless the drug is intended for the treatment of malignancies (i.e. oncology).
In paediatric clinical trials, invasive procedures are avoided or at least minimised for ethical reasons and the usefulness of clinical trials in children has been widely debated over the last decades, 1 as highlighted by two papers recently published in the journal of the American Academy for Paediatrics.2,3 Several authors and specialists have reported a critical need for more clinical studies in paediatrics combined with an improvement in the methodologies used in practice. Some authors have argued that incorporating prior knowledge from adults should help to attain a better understanding of the paediatric population. However, other studies have shown that children should not be considered small adults but rather a specific population with a different metabolism that is not necessarily linearly related to growth.1,4
For dose-finding paediatric studies, guidelines have been suggested for the choice of starting subset doses 5 (e.g. the starting dose should equal 80% of the adult recommended dose, and these doses should then be increased by 30% to obtain the subset doses). However, these recommendations are arbitrary and do not rely on any scientific justifications. As a result, to improve the selection of the dose-range that should be used in a paediatric study based on the use of adult information, this information should be investigated through (1) the choice of the dose-range for a paediatric trial, (2) the dose-finding model and (3) its parameterisation.
In the development of a dose-finding model for the paediatric population, difficulties regarding the evaluation of toxicity alone (except in oncology) have led to the use of a joint model for both toxicity and efficacy instead of a model that evaluates toxicity prior to efficacy. Several statistical methods are available for the design of early stage phase I/II clinical trials. Among them, Bayesian methods, such as the EFFTOX design and the bivariate Continual Reassessment Method (bCRM), have been proposed.15,16 Although initially used in oncology settings, these methods have also been used for studies of the paediatric population. 17 Additionally, Broglio et al. 18 proposed a method in which adult and paediatric trials are performed simultaneously with dose-finding models for each population that share an identical slope but a different intercept. Doussau et al. 5 reviewed the methods that could also be used in paediatrics, such as ‘3+3’, CRM with its modifications and Dose-finding with Escalation with Overdose Control (EWOC).
The use of an adaptive dose-finding method requires that three components be fixed prior to initiation of the trial:
The aim of this paper is to propose a unified approach for the design of a paediatric dose-finding clinical trial through the extrapolation and bridging of information gleamed from the adult population. We have gathered and modified various methods that have been developed in different fields to propose a unified approach. The novelty of our work consists of the proposal of extrapolation with maturation from adult PK into the definition of the dose-range (1) and of the use of adult information from several sources to better parameterise the dose-finding designs (2) to (3) instead of leaving these decisions to arbitrary choices. In this work, several options are proposed for the selection of the dose-range, the WM and/or the parametrisation of the dose-finding design (Figure 1). Section 2 details the dose-finding model, illustrates the options for specifying the dose-range and describes the parametrisation of the design using adult information. The simulation settings and results are given in Sections 3 and 4. Finally, the guidelines are proposed in Section 5 and a discussion is provided in Section 6.
General framework describing the different proposed steps in the planification of paediatric dose-finding clinical trials. It is composed of (1) the choice of the dose-range with three different possible options, linear adjustment (LA), allometric adjustment (AA) and maturation adjustment (MA). They are built using extrapolation from adults to children, with di the paediatric dose, ci the adult dose and Wch and Wad, respectively, the children and adult weight; (2) the working model (WM) specification, where adult PK and toxicities can be used to built a toxicity function η. It allows to calculate the WMs (αi, 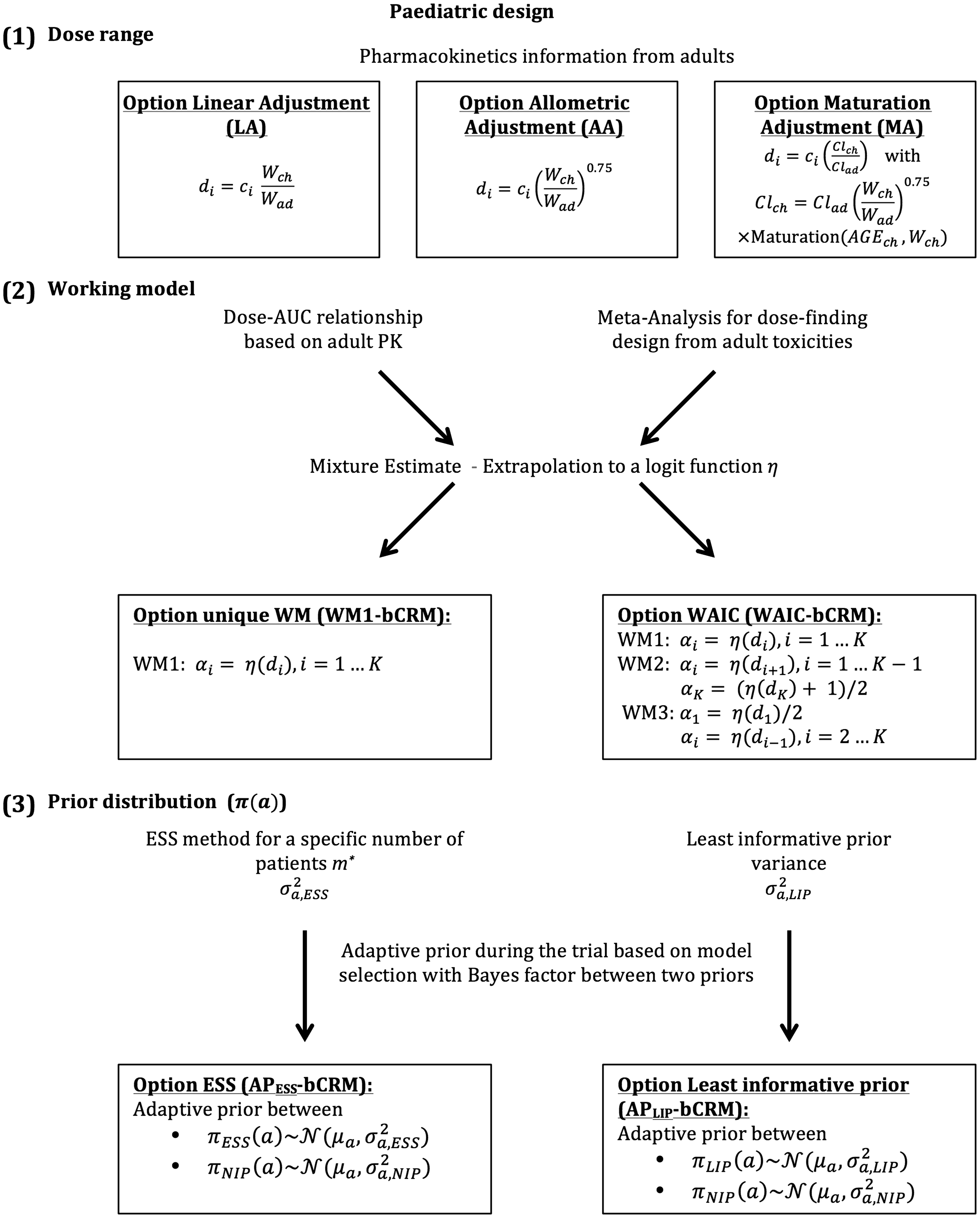
2 Model and methods
We considered the design of a phase I/II clinical trial in the paediatric population using the Bayesian bCRM as the dose allocation method. Section 2.1 presents the bCRM method and the dose allocation algorithm. The first step (1) consists of defining the doses to evaluate. We proposed three options for the selection of the dose-range using adult to paediatric extrapolation methods, which use different adjustments of the paediatric dose from the adult’s recommended dose: linear, related to weight with allometry and related to physiological processes with maturation functions to account for maturation differences between adults and children. These three options are described in Section 2.2.1. Once the doses are defined, step (2) consists of associating each dose with a given initial guess of the toxicity and efficacy probability, and these relationships are called ‘working models’ (WMs). The selected doses are supposed to be within a desirable toxicity, and efficacy interval to ensure that patients are not overtreated or undertreated. The WMs are constructed by gathering several prior sources of information from the adult population, such as PK, phase I trials, phase II trials, toxicity and clinical response. We proposed two options for the WMs: using only one WM, or using several WMs and selecting the optimal WM using automatic criteria. A description of the methods used to elaborate the WMs is given in Section 2.2.2. Finally, step (3) involves the selection of the dose–response parameter density of the priors used in the bCRM. We proposed two options for these priors: considering adult information or considering the case with the least information. These are described in Section 2.2.3, and a summary of this general framework is presented in Figure 1.
2.1 Bivariate continual reassessment method (bCRM)
In this general framework, we used the bCRM as phase I/II dose-finding methods. This design proposes a joint model for both toxicity and efficacy.16,28 The aim of this method is to identify the safe most successful dose (sMSD), which is the most successful dose under toxicity restriction. Let
Following the under-parametrised model approximation proposed by O’Quigley et al.,
28
we have
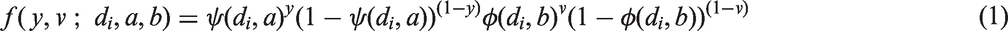
Under Bayesian inference, the prior distributions for a and b are respectively denoted by
The dose allocation rule is the following. Let us denote
2.2 Extrapolation from adult data to paediatrics
Similarly to any model-based phase I/II dose-finding method, the design can be sensitive to three settings: (1) the choice of dose-range, (2) the WMs and (3) the prior distributions. In our proposed method, we suggest that these settings be based on extrapolations from the adult to paediatric population.
2.2.1 Specification of a dose-range
Paediatric data are often rare, and paediatric doses are usually selected based on existing recommendations for adult doses. We proposed three options for the selection of paediatric doses: linear and allometric extrapolation from adult doses, which are the current practices, and use of maturation, which is a novel approach in this context.


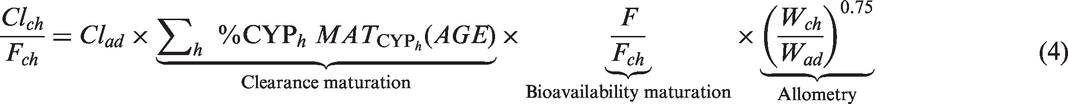
Using allometry to account for size, the bioavailability and clearance sections of the equation account for the maturation process in the paediatric population.4,29 The maturation of clearance depends on cytochromes (CYPs), which are responsible for the hepatic elimination process. In equation (4),
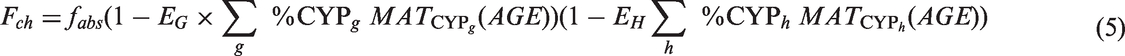
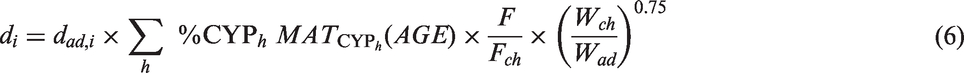
The three above-described options were compared by building a dose-range for LA, allometric adjustment (AA) and MA. The adult average weight Wad was considered to equal 70 kg, and the average paediatric weight is not properly defined. A population of N = 100,000 patients aged 0 to 21 years was then simulated using P
3
M software30,31 and for each simulated subject, the individual clearance
2.2.2 Choice of WMs using adult information
After selecting the dose-range for the study, the next step is to parametrise the model-based dose-finding method, i.e. the bCRM. In this method, the WMs αi and
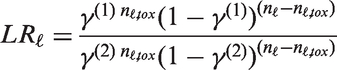
We now describe in detail the two proposed options.

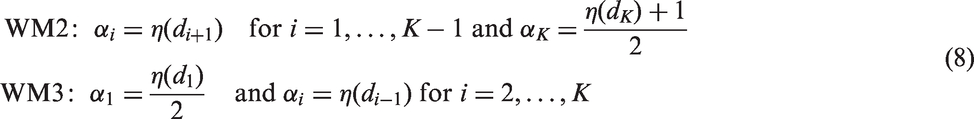
The bCRM was performed for the three WMs, and the model selection was based on the WAIC22,23 was applied. This approach selected the WM that best fit the data and returned an estimate of parameters a and b for each dose i.
2.2.3 Specification of prior density
In addition to the WMs, when using Bayesian model-based methods, the prior density of the dose–response model needs to be specified. In our framework, the prior distributions of the dose–toxicity model parameters were selected using two different parametrisations based on either (i) the adult information (option ESS, AP ESS -bCRM) or (ii) least information (option least informative prior, AP LIP -bCRM). In the first option, due to the sparsity of the data, it appears appropriate to attempt to incorporate observations into the prior. However, the information introduced by the prior distributions to the posterior should not overtake the information introduced by the likelihood distribution.
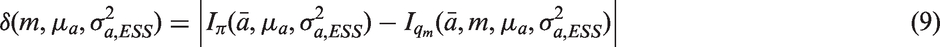
However, the resulting variances
The decision to switch from
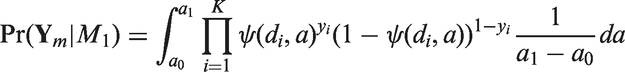
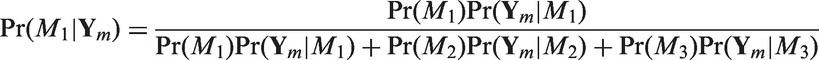
In practice, a comparison was performed between AP
ESS
-bCRM, which used the bCRM with the adaptive prior from
3 Simulations
The aim of the simulation study was to evaluate and compare the performances of each dose-range and model setting proposition, in terms of selected dose. Based on the motivating illustration, we proposed to plan, conduct and analyse a hypothetical phase I/II dose-finding clinical trial for erlotinib in the paediatric population. We used PK parameters as well as dose-finding toxicity and efficacy clinical trial data for erlotinib obtained from the adult population for extrapolation and bridging.
Model settings for simulations. AP
ESS
-bCMR uses adaptive prior from Toxicity, efficacy outcomes and the number of treated patients of erlotinib treatment. Note: Toxicities are skin rash of grade 3 or more and efficacy, defined as stable disease and above (RECIST), was limited to glioblastoma. The distributions for calculating the mixture Representation of the estimated probabilities of toxicity used to build WMs for paediatrics according to dose (mg/kg). The logit function


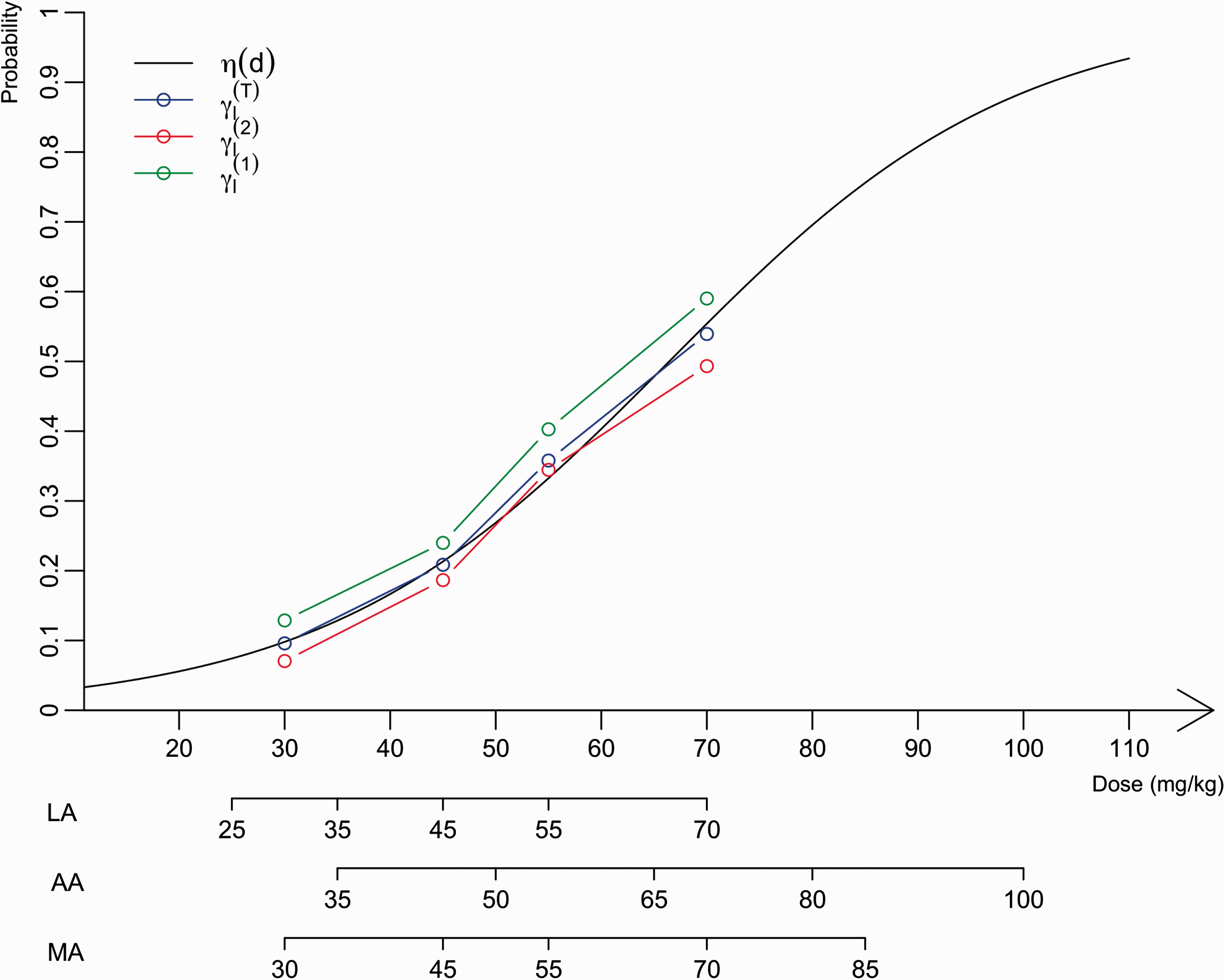
For efficacy, the data from adults treated for glioblastoma were considered because efficacy is strongly related to the specific disease. In this setting, efficacy was defined as remission or stability regarding tumour size according to RECIST criteria. Because most of the data were associated with one dose, a method developed by Cheung et al.
35
was used to obtain the WM. The percentage of efficacy over all available published data (Table 2) was 20%. We obtained the WM for efficacy reported in Table 1 using the function getprior(halfwidth
With the ESS option, μa and
Then,
For efficacy, prior
The performances of our unified approach were investigated through a simulation study under several scenarios presented in Figure 3 for the three dose-ranges options (LA, AA and MA). Extrapolation from adults yielded an initial estimate of 48 mg/kg for the MTD associated with a toxicity target of 0.25. We aimed to evaluate how this choice influences the performance of our proposed methods by selecting scenarios in which the MTD and sMSD were different. Scenarios 1, 2 and 3 were based on the results of two real paediatric trials conducted by Geoerger et al.
14
and Jakacki et al.
15
For all three scenarios, we considered the same MTD that was found in each trial, and the efficacy was simulated. In scenarios 1 and 2, the MTD (83 mg/kg) is equal to that reported by Geoerger et al. and is far from the efficacy extrapolated from adult information (48 mg/kg). In scenario 1, the sMSD was similar to the MTD, whereas in scenario 2, the sMSD was 65 mg/kg. In scenario 3, the MTD and the sMSD are equal to those reported by Jakacki et al. (55 mg/kg) and close to the value extrapolated from adult information. Finally, we added three scenarios: in scenario 4, the MTD (65 mg/kg) was equivalent to the MSD; in scenario 5, the MSD was higher than the MTD (45 mg/kg) and in scenario 6, the sMSD is similar to the MTD (70 mg/kg).
Presentation of the six scenarios used in the simulation study. The dose–toxicity, R(d), curve is in red, the dose–efficacy, Q(d), curve is in blue and the dose-success, P(d), curve is in green. The sMSD is represented by black vertical line, the toxicity and efficacy targets are given with dashed lines. The admissible doses (AD) are given by the green area under the success P(d) curve.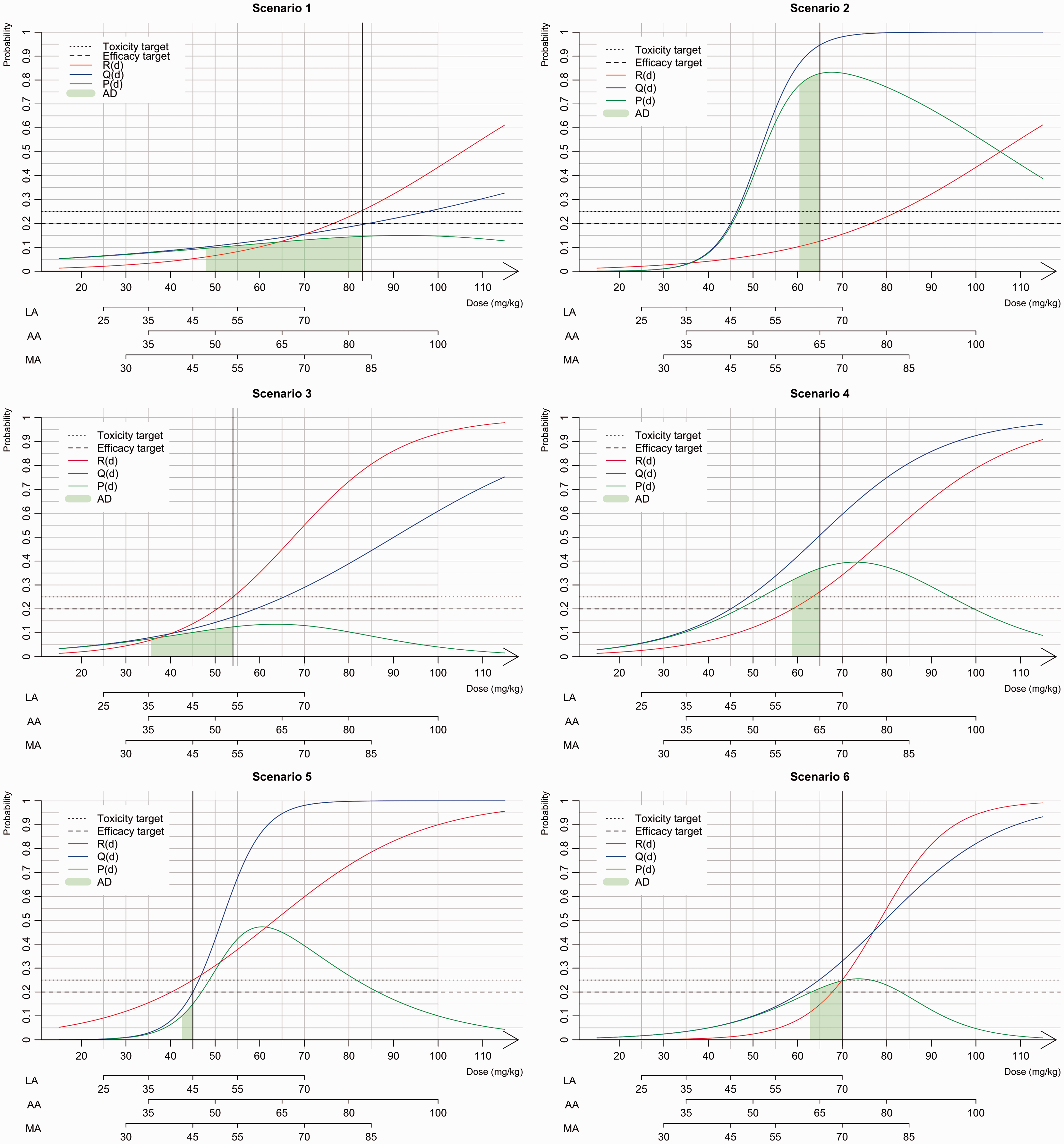
For each scenario, we performed 1000 simulated phase I/II trials with a maximal sample size of N = 50 patients. Because maturation is known to differ among different paediatric age subgroups, we selected a paediatric population with an age range of 2 to 5 years. We also chose a toxicity target of
For each approach, the percentage of correct dose selection (PCS) of the sMSD was computed. We also evaluated the percentage of acceptable doses (ADs) that includes the closest dose to the sMSD for each approach; if this dose existed, we evaluated the next lower dose for which the probability of success P was included in
4 Results
Based on the toxicity results reported by Geoerger et al., 13 scenarios 1 and 2 shared the same MTD of 83 mg/kg. However, the sMSDs differed depending on the efficacy differed with 83 mg/kg for scenario 1 and 65 mg/kg for scenario 2 (Figure 3).
Simulation study results for the three dose-range methods, linear, allometry and maturation adjustment (LA, AA, MA) under several scenarios.

Note: The percentage of correct selection (PCS) are represented in italic, that is, the sMSD. The percentage of acceptable dose (underlined) are summed up in bold. The simulation setting for each approach are given in Table 1.
In scenario 3, the sMSD was equal to the MTD (i.e. the 54 mg/kg dose). In the case of AA, the closest dose to the MTD was 50 mg/kg, and the PCSs for all options were greater than 71%.
In scenario 4, the sMSD and MTD were similar (the 65 mg/kg dose). In the case of AA, the dose was within the dose-range, and the PCSs of WAIC-bCRM and WM1-bCRM were 70.5% and 75.2%, respectively. However, the AP ESS -bCRM gave a lower PCS (63.9%) compared with that obtained with the AP LIP -bCRM (73.6%). In scenario 5, the recommended dose was 45 mg/kg, which is within the dose-range obtained with LA and MA. In this case, all options gave high PCS values greater than 60%. If the dose was not within the range, as was the case with AA, the PCS decreased an average of 10%. In scenario 6, the recommended dose was 70 mg/kg. Even if the dose was only in the ranges obtained with the LA and MA options, high PCS values (above 90%) were obtained for all dose-range options.
The comparison of the performances of AP ESS -CRM and AP LIP -CRM, revealed similar performances over all dose-range options and scenarios. However, WM1-bCRM and WAIC-bCRM generally provided better recommendations in terms of the admissible dose.
In the case of a too-toxic scenario (sMSD of 20 mg/kg, data not shown), the stopping rules allowed the trial to be stopped, if a toxic reaction was observed in 90% of the cases, regardless of the method.
In general, if the sMSD was within the dose-range, the PCS and AD percentages were high, whereas if the dose was close but not within the range, a lower PCS percentage and a rather high AD percentage were obtained.
5 Guidelines
Based on the results of our simulations, we suggest the following settings for the proposed approach:
For dose-range selection: use either options AA or MA. For the WM choice: use option WAIC-bCRM because our results indicate that it is better to use several WMs in the model selection process than a unique WM. For prior distribution: if the quantity and quality of the adult information is high, use the AP
ESS
-bCRM option; however, if there is some doubt regarding the available adult information, use the AP
LIP
-bCRM option.
6 Discussion
In this work, we present a unified approach for planning, conducting and analysing paediatric dose-finding clinical trials. This unified approach is based on several possible methods that aim to improve the choices made in the design of paediatric trials. For the analysis of the paediatric population, for which only a small number of clinical trials have been conducted and which typically includes a small number of patients, the bridging of information from the adult population (when possible) to the paediatric population, particularly using PK extrapolation tools such as allometry and maturation functions, is highly relevant.
We based our unified method on the bCRM, which jointly models toxicity and efficacy with a dose-finding allocation rule because in paediatric populations, safety takes priority over efficacy. Our unified approach includes all stages in the dose-finding process, ranging from dose-range selection to the choice of prior distributions for dose responses.
The first step of our work proposed three different dose-range adjustments (i.e. LA, AA or MA). The resulting dose-ranges overlapped, and a wider range was obtained with AA. In this study, we used the specific context of erlotinib, a drug that has been investigated in both adult and paediatric populations for cancer treatment. Both dose-finding and PK studies in adults and children are available. We thus used the available adult information to plan a paediatric trial using the proposed extrapolation and bridging methods and used the children’s dose-finding data to build scenarios for the simulation study, which allowed us to evaluate our design choices.
Our extrapolation and bridging approach used data from more than 580 adult observations. We based three of our scenarios for the simulation study on the toxicity observations reported by Geoerger et al. 13 and Jakacki et al., 14 who performed trials that evaluated 16 and 19 children, respectively. Thus, the estimation of the MTD or recommended dose in each trial was associated with high variability due to the small sample size. In this case, it is difficult to assess how far from reality is our model from the true paediatric population. In general, our results show that in cases in which the MTD and sMSD are far from our initial guess (as in scenarios 1 and 2), our proposed dose-finding designs based on either model selection criteria or adaptive priors performed well. A similar finding was obtained for scenario 3, in which the MTD and the sMSD were not far from our initial guess. These results are in favour of the implemented methods because misspecified initial choices do not impact the performance of our proposition.
To date, there is no clear recommendation for the selection of the dose-range that should be used in paediatric dose-finding clinical trials. Allometric scaling was initially introduced by West et al. 36 for identifying measurements that work across and within species. Several studies have suggested that the allometric coefficient may be different in early childhood.36,37 The discrepancy between size-based scaling and effective changes in paediatric patients, particularly neonates and infants, can also be explained by differences in physiological processes due to maturation.
The second step of our work was to propose dose-finding design choices for the dose allocation process using adult clinical trial observations. Because not all of the calculated doses were used for adults, we needed to build a logit function based on mixture estimates in adults. For this purpose, we assumed that the exposure was similar in both adults and children. Adult PK combined with maturation served as the first source of information for the toxicity probability, which was defined in terms of PK (AUC or Cmax). A direct curve was reported by Thomas et al. 34 The second source of information was toxicity from early-phase clinical trials in adults. This method allowed us to propose tools for the establishment of the WMs and for the prior distributions of dose–toxicity parameters.
For simplicity reasons, we maintained the same scenarios for all dose-ranges, which led to different sMSDs. In cases in which the model hesitated between two doses, a lower PCS was obtained primarily because the real dose was not exactly within the dose-range. Other scenario choices could have favoured one adjustment method over the other, although this situation occurred due to arbitrary choices. Other methods that jointly model toxicity and efficacy for dose-finding, such as EFFTOX, can also benefit from our proposed approach, although some may only need to use part of our model. 16 In our case, power function modelling of the dose–toxicity or dose–efficacy curves was selected for simplicity. However, several other models, such as the logit model, could easily be used in our setting.
In conclusion, the bridging and extrapolation of adult data for the design of paediatric dose-finding clinical trials appeared to improve the results of these studies. Our proposition may prove helpful for physicians and statisticians who wish to plan and conduct early-phase trials in this population. We attempted to unify and modify existing methods to obtain a clear stream of decision making regarding several crucial choices that need to be made prior to initiation of a trial. We believe that this approach will improve and allow better use of the available information sources for the planning of new trials.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Caroline Petit was supported during this work by a grant IDEX from the Université Sorbonne Paris Cité (2013, project 24). Sarah Zohar, Emmanuelle Comets and Moreno Ursino were funded by the InSPiRe (Innovative Methodology for Small Populations Research) Project of the European Union Seventh Framework Programme for Research, Technological Development, and Demonstration under grant agreement FP HEALTH 2013-602144. During this work, Adeline Samson was partially supported by the LabEx PERSYVAL-Lab (ANR-11-LABX-0025-01) funded by the French program Investissement d’avenir.