
Abstract
Lymphoid interstitial pneumonia (LIP) is a rare form of interstitial pulmonary disease, which has been described in association with a wide range of autoimmune disorders. Although the association of this entity with Sjogren’s syndrome is well known, only a few cases are reported in relation to systemic lupus erythematosus (SLE). The aim of this paper is to review the cases reported in literature to date, as well as to describe the characteristics of these patients including the new case presented herein. We will be focusing on the case of a 36-year-old female patient diagnosed with SLE on hydroxychloroquine treatment who develops pleuritic chest pain and progressive dyspnea after 3 years of follow-up. The chest CT scan showed pleural thickening and both multiple and bilateral micronodules. A lung biopsy was also performed, revealing an infiltration of lymphocytes, plasma cells, and histiocytes in the alveolar septa suggestive of LIP. After conducting a review of the literature, we identified seven other cases describing SLE in association with LIP. The majority of them were young women, and LIP tends to appear early in the course of the disease, even as a form of initial presentation in some cases. Symptoms included cough, dyspnea, and pleuritic pain, with the exception of one case which was asymptomatic. It is noteworthy that half of the patients were positive for anti-SSA/anti-SSB autoantibodies, and some of them also met criteria for Sjogren’s syndrome. Treatment with steroids and other immunosuppressive agents improved symptoms in all of them.
Keywords
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease with a great variety of clinical manifestations that can affect many organ systems. Even though clinical presentation can be highly variable, pulmonary involvement is usually not present. 1 Lymphoid interstitial pneumonia (LIP) is a rare form of interstitial lung disease, within the spectrum of benign pulmonary lymphoproliferative disorders, characterized by a dense polyclonal interstitial infiltration of lymphoplasmacytic cells. 2 It has been described in association with infections, such as HIV or Epstein–Barr virus, and especially in autoimmune disorders, among which the association with Sjogren’s syndrome (SS) is the most well-described.3,4 We herein present a clinical case report of LIP as a form of lung involvement in a patient with SLE and a review of the literature to date. Verbal consent was obtained from the patient for this case report.
Case presentation
We present the case of a 36-year-old woman, with a history of thymectomy with no data of recurrence, who was under follow-up by our Systemic Diseases Unit since 2019, with a diagnosis of SLE with articular affection as the predominant manifestation. She had positive antinuclear antibodies (homogeneous pattern 1/320) and double-stranded DNA, being negative for anti-SSA/anti-SSB autoantibodies. She was on treatment with hydroxychloroquine with an SLEDAI of 0 points.
In March 2022, she reported fever and chest pain. Initially, she was diagnosed with respiratory infection and treated with both moxifloxacin and prednisone, with a good response. A thoracic CT scan was also performed (requested in a routine oncology check-up), which revealed multiple and bilateral pulmonary micronodules, some of them even cavitated. Given this scenario, an interferon-gamma release assay (IGRA) was requested to rule out tuberculosis, with negative result, and a bronchoscopy was performed without any pathological findings. As a first possibility, these results could suggest necrosis secondary to vasculitis in the context of SLE.
Later in May, she attended outpatient clinics referring a worsening of her general condition, with the appearance of dyspnea upon exercise, pleuritic chest pain, and occasional palpebral ptosis. In view of this situation, the patient was admitted to hospital for further study. Her vital signs were normal, including respiratory rate and oxygen saturation. Physical examination was unremarkable. Laboratory tests showed mild normocytic anemia and scarce increase in PCR. Glucose, electrolytes, complement, renal, and liver functions were normal. She was also evaluated by neurology with the diagnosis of myasthenia gravis. A chest CT scan was requested, which revealed the persistence of multiple nodules in both lungs and pleural thickening with a basal predominance, without pleural effusion, not shown in previous images (Figure 1(a)). A PET-CT scan was also performed, with evidence of this bilateral pleural thickening (left SULmax 7.28, right SULmax 4.21) and pulmonary nodules with low metabolic uptake (Figure 1(b)). As these findings suggested neoplastic/metastatic affection in first place, a pleural biopsy was carried out, but it could not be analyzed due to an insufficient sample. (a) CT scan showing pulmonary nodules and pleural thickening. (b) PET-TC demonstrating pulmonary nodules with low metabolic uptake.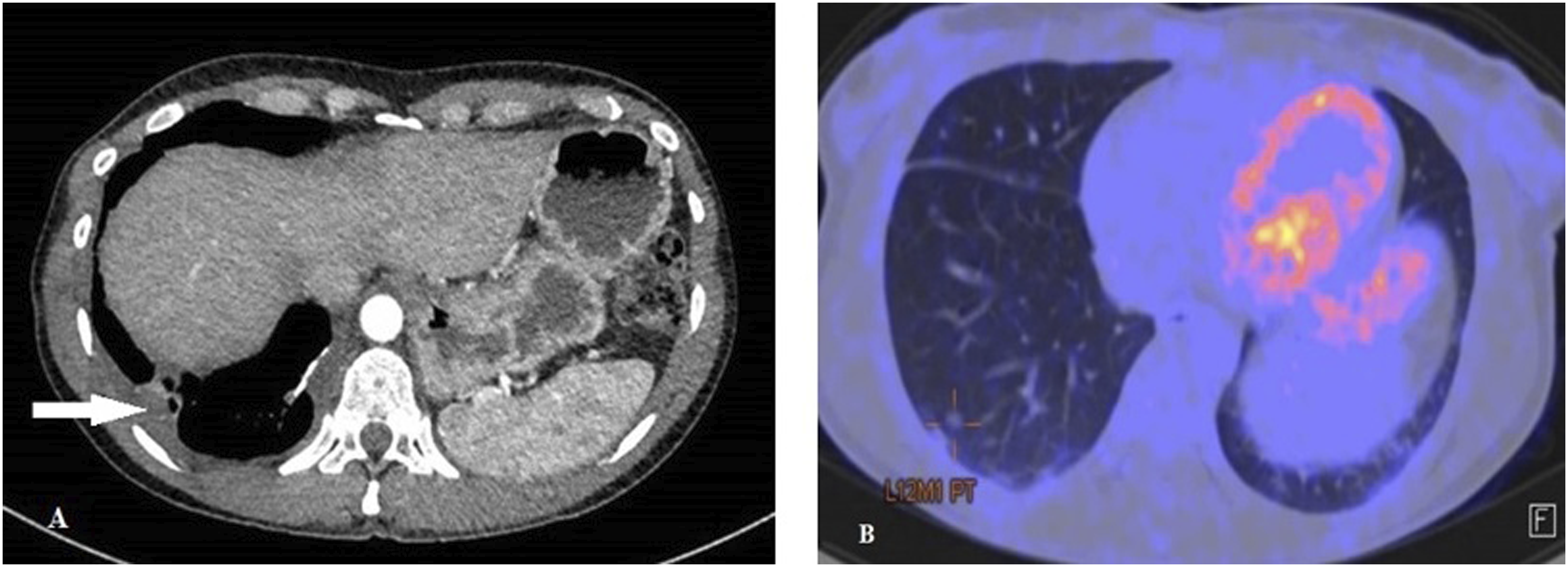
Upon discussion with the thoracic surgical committee, a surgical lung biopsy was finally performed, revealing polyclonal infiltration of lymphocytes and plasma cells around bronchioles and alveolar septa, with no evidence of organisms, granuloma, or vasculitis, being also histologically negative for IgG4-related disease (Figure 2(a) and 2(b)). These pathologic findings, in relation to the context of the patient’s disease, were suggestive of LIP. Treatment with oral prednisone (30 mg daily) and azathioprine was started with adequate initial tolerance and improvement. The patient’s evolution is still pending and will be assessed by follow-up in our Systemic Diseases Unit. (a) Lung biopsy showing dense infiltrate of lymphoplasmacytic cells. Hematoxylin eosin ×10. (b) Lung biopsy demonstrating the presence of lymphoid follicles. Immunoperoxidase ×2.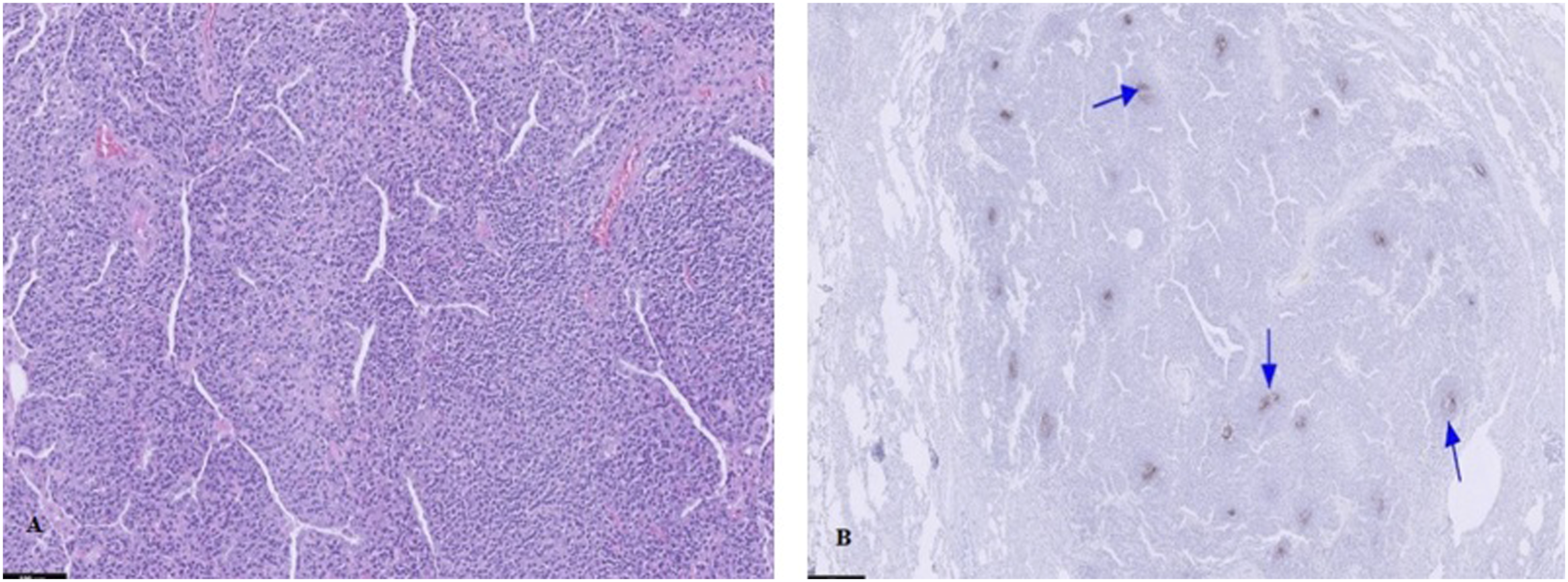
Methods
A literature research of case reports about LSE in association with LIP was performed in the PubMed database. Systemic lupus erythematosus, lymphoid interstitial pneumonia, lymphocytic interstitial pneumonia, and lymphoid interstitial pneumonitis were the key words used to find case reports. Some articles in which SLE was not the main diagnosis (overlapped with other autoimmune disorders) were excluded. Eventually, the research was concluded with seven cases meeting these criteria. A descriptive analysis of baseline characteristics, laboratory data, imaging tests, treatment received, and outcomes was performed.
For this purpose, a statistical analysis was conducted. Results are expressed as percentages for dichotomous and non-dichotomous qualitative variables, and continuous variables are usually distributed as mean +/- standard deviation (SD). Missing data were excluded.
Results
Characteristics of the patients with LIP in association with SLE.
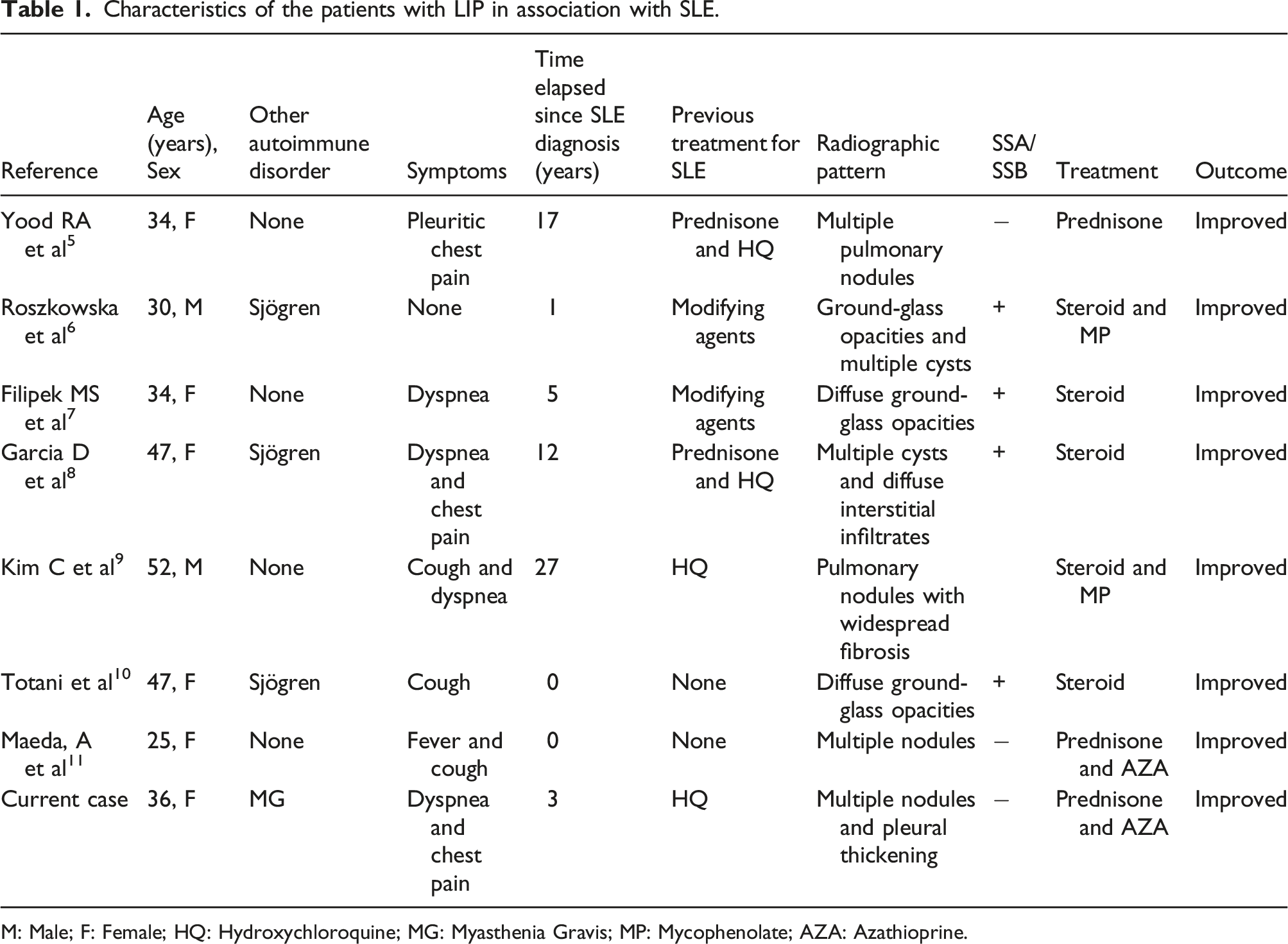
M: Male; F: Female; HQ: Hydroxychloroquine; MG: Myasthenia Gravis; MP: Mycophenolate; AZA: Azathioprine.
Discussion
Pulmonary involvement in SLE is not often present, with pleuritis being the most frequent manifestation occurring in a 16–60% of the patients. 12 Compared to other systemic diseases such as rheumatoid arthritis, systemic sclerosis, or dermatomyositis, lung involvement is unusual in SLE. The prevalence of interstitial lung diseases associated with SLE is about 3–9%.13,14 Within these infrequent entities, LIP is a rare form, only found in a few cases in the literature,5–11 while the association between LIP and SS is the most described. Several studies have pointed out that the relation of SLE and SS may increase the prevalence of interstitial lung disease.3,14
The pathogenesis of LIP is still poorly understood. It is considered part of the spectrum of benign lymphoproliferative disorders that can affect the lung, and it is taught to be triggered by multiple stimuli such as infections like HIV or EBV. However, its strong association with autoimmune disorders suggests that LIP itself may be considered an autoimmune disease.13–15 It is estimated that nearly 40% of the cases occur in the setting of an autoimmune process, with approximately 25% of the cases described in association with SS. 14 Other autoimmune associations with a much lower incidence—in which SLE is described—include rheumatoid arthritis, myasthenia gravis, pernicious anemia, Hashimoto’s thyroiditis, celiac sprue, or primary biliary cirrhosis.
Clinical manifestations of LIP include cough, dyspnea and, less frequently, asthenia, weight loss, and pleuritic chest pain.15,16 The radiological presentation is not specific and can be highly variable. It usually includes reticular or reticulonodular opacities with predominance of the lower lung area. Pulmonary cysts may be observed in up to 80% of patients with LIP and may help differentiate LIP from lymphoma. 17
Diagnosis requires a histological lung sample with demonstration of polyclonal lymphocytic infiltration, plasma cells, and histiocytes in alveolar septa. 15 The treatment for LIP is not well established yet. Glucocorticoids and other immunosuppressive agents, such as azathioprine, cyclosporine, or cyclophosphamide, have been used with different results.15,16 Prognosis is highly variable as well. Some patients may improve even without treatment, while others may progress to advanced interstitial fibrosis. Generally, the 5-year mortality remains between 33% and 50%, and it could likely vary depending on the underlying disease process. It is estimated that 5% of the cases may transform into pulmonary lymphoma. Causes of death include intercurrent infection and end-stage pulmonary fibrosis. Moreover, an increase of death from other non-pulmonary causes has also been observed.15,16,18
Our review suggests that the form of presentation, imaging tests, and therapeutic approach in patients with SLE does not differ much in relation to the rest of the cases with LIP (idiopathic or with other associations) described in literature. The trend is that LIP tends to appear mostly in young women with stable or initial phase of SLE. Also noteworthy is the association with other autoimmune disorders, especially SS, and some laboratory characteristics such as antiRo/La autoantibody positivity. Our sample size is too limited to draw conclusions, but it seems that the association of SLE with other autoimmune disorders, SS in the first place, may increase the risk of developing this type of lymphoproliferative disorder of the lung.
Since the development of LIP in patients with SLE is very uncommon, more data are required in order to better understand this association and the prognosis of the disease in these patients. The reviews found in literature give an overview of the published cases with LIP by analyzing morbimortality in general lines. Since the number of cases with LIP is limited and their associations highly variable, no morbimortality studies differentiating the underlying cause has been performed to date. Among the case reports analyzed in this essay, it seems that treatment improved symptoms in all patients, but it would be interesting to have more data on long-term follow-up to see if there really is a remission of the disease or, on the contrary, it progresses or results in some other outcome.
Conclusion
This review suggests that LIP may appear as an unusual form of interstitial pulmonary affection in patients with SLE. Clinicians must be aware of this entity, and it should be included in the differential diagnosis of patients affected with SLE who develop respiratory symptoms, especially when observed in young women, not late in the course of the disease, and even when symptoms appear in the setting of SLE. The presence of concomitant or secondary SS, SSA/SSB autoantibody positivity or the appearance of multiple micronodules or cysts in chest CT images could suggest this etiology, but definitive diagnosis will be achieved with a lung biopsy confirming the histological pattern of LIP. Steroid therapy in combination with other immunosuppressive agents, such as azathioprine and mycophenolate, seems to improve symptoms in these patients. Further studies are needed for a better understanding of the course of the disease and prognosis in these patients.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.