
Abstract
Background
Systemic lupus erythematosus (SLE) disproportionately affects individuals of African ancestry (AA) compared to European ancestry (EA). In the general population, high risk (HR) variants in the apolipoprotein L1 (APOL1) gene increase the risk of renal and hypertensive disorders in individuals of AA. Since SLE is characterized by an interferon signature and APOL1 expression is driven by interferon, we examined the hypothesis that APOL1 HR genotypes predominantly drive higher rates of renal and hypertensive-related comorbidities observed in SLE patients of AA versus those of EA.
Methods
We performed a retrospective cohort study in patients with SLE of EA and AA using a genetic biobank linked to de-identified electronic health records. APOL1 HR genotypes were defined as G1/G1, G2/G2, or G1/G2 and low risk (LR) genotypes as 1 or 0 copies of the G1 and G2 alleles. To identify renal and hypertensive-related disorders that differed in prevalence by ancestry, we used a phenome-wide association approach. We then used logistic regression to compare the prevalence of renal and hypertensive-related disorders in EA and AA patients, both including and excluding patients with the APOL1 HR genotype. In a sensitivity analysis, we examined the association of end stage renal disease secondary to lupus nephritis (LN-related ESRD) with ancestry and the APOL1 genotype.
Results
We studied 784 patients with SLE; 195 (24.9%) were of AA, of whom 27 (13.8%) had APOL1 HR genotypes. Eighteen renal and hypertensive-related phenotypes were more common in AA than EA patients (p-value ≤ 1.4E-4). All phenotypes remained significantly different after exclusion of patients with APOL1 HR genotypes, and most point odds ratios (ORs) decreased only slightly. Even among ORs with the greatest decrease, risk for AA patients without the APOL1 HR genotype remained significantly elevated compared to EA patients. In the sensitivity analysis, LN-related ESRD was more prevalent in SLE patients of AA versus EA and AA patients with the APOL1 HR genotype versus LR (p-value < .05 for both).
Conclusion
The higher prevalence of renal and hypertensive disorders in SLE patients of AA compared to those of EA is not fully explained by the presence of APOL1 high risk variants.
Systemic lupus erythematosus (SLE) is an autoimmune disorder that disproportionately affects individuals of African ancestry (AA) compared to those of European ancestry (EA). 1 In the United States, patients of AA have a two- to three-fold increase in the prevalence and risk of SLE. 2 Patients of AA also experience more severe disease, with higher rates of lupus nephritis (LN) 3 and a 3-fold increased risk for end-stage renal disease (ESRD) compared to patients of EA. 4 Previous research has used a phenome-wide association study (PheWAS) approach to detect differences in the prevalence of certain phenotypes among SLE patients of AA and EA; many of these phenotypes were related to renal disease or its complications. 5 Additionally, hypertension is more frequent in SLE patients of AA, and the prevalence of resistant hypertension is almost twice that observed in patients of EA, 6 although these discrepancies are also observable in the general population.7,8
SLE is a highly heritable disease, 9 and while trans-ancestry studies have shown that risk alleles for SLE are shared among different genetic ancestries, 2 fine mapping approaches have also identified novel risk alleles that have different frequencies among SLE subpopulations. 10 These findings may explain observed phenotypic differences by ancestry in patients with SLE. 11 For example, the prevalence and time to progression to ESRD in patients with LN are both associated with the presence of APOL1 high risk (HR) genotypes, 12 genotypes that are absent in individual of EA ancestry. 13 Prior research has further determined that the APOL1 HR variants are a major contributor to the excess risk for kidney diseases in the general population of individuals of AA (current estimates suggest approximately 70% of excess risk).14–16 Moreover, individuals with these HR genotypes have higher systolic blood pressure and earlier diagnosis of hypertension than those who do not. 20 These associations have made APOL1 HR genotypes an appealing potential target for treatment.
APOL1 is a gene located on chromosome 22, 17 with two alleles, (G1 and G2) that underwent evolutionary selection in African populations because they confer increased resistance to Trypanosoma brucei; 18 however, individuals carrying two alleles have a marked increase in the risk of renal disease. The G1 allele is a set of two mutations (rs73885319 and rs60919145) near the C terminal that are in complete linkage disequilibrium (LD) in individuals of AA in the US; 19 the G2 allele is a six base pair deletion (rs71785313) in the same functional domain of APOL1. 18 High risk genotypes for APOL1 are defined as homozygotes for either G1 or G2 (G1/G1, G2/G2) or compound heterozygotes (G1/G2). Approximately half of African American patients carry either the G1 or G2 allele and approximately 12%–15% carry two alleles 18 (i.e., an HR genotype).
APOL1 transcription is enhanced by inflammatory factors that include those activated through interferon-dependent pathways. 21 SLE characteristically activates the interferon system, 22 increasing the expression of interferon-regulated genes. Concordant with these mechanisms and as noted above, APOL1 HR genotypes are associated with ESRD as a complication of LN.23,24 However, it is not known if the increased risk of renal disease and its complications in SLE patients of AA compared to those of EA is explained by the APOL1 HR genotype. 3 The determination of this association is important because several drugs are in development to target APOL1 and thus could have potential use in patients with SLE.21,25 Therefore, we examined the hypothesis that APOL1 HR genotypes predominantly explain the increased prevalence of renal and hypertensive comorbidities in SLE patients of AA compared to those of EA.
Materials and Methods
We performed a retrospective cohort study among patients of EA and AA diagnosed with SLE who had genotyping data available in BioVU, a genetic database linked to de-identified electronic health records (EHRs) at Vanderbilt University Medical Center (VUMC).26,27 EHR data included medications, laboratory results, demographics, and diagnostic codes. The study was approved by the VUMC Institutional Review Board and was classified as non-human subjects research.
Study population: The cohort included 784 patients with SLE, defined as a patient who was diagnosed as having SLE by a rheumatologist, nephrologist, or dermatologist and confirmed by the investigators. Potential cases of SLE were selected using validated algorithms 28 to detect likely cases of SLE in the EHR, followed by investigator-performed chart review to confirm the diagnosis of SLE. The algorithms included antinuclear antibodies titers, diagnostic codes, and mention of combinations of medications such as hydroxychloroquine and immunosuppressants. 28 LN was defined as the presence of (a) biopsy-proven renal disease attributed to SLE; 29 (b) ESRD attributed to SLE (i.e., no other conditions could explain it) without kidney biopsy; (c) proteinuria (>0.5 gm/day) or 3+ on urinalysis, or (d) cellular casts. 30 Follow-up began at the date of the first EHR and ended at the first of death, lost to follow-up, or end of the study (02-22-2022).
Outcomes: Disease diagnoses for the primary analysis were defined using the International Classification of Diseases Ninth and Tenth Revision (ICD9/ICD10) diagnosis codes recorded in the EHR through the end of follow-up. The ICD9/ICD10 codes were mapped to phecodes31,32 to assess the prevalence of more than 350 conditions which met the criteria for a PheWAS, as described below. Given that phecodes may lack precision for particularly complex diagnoses (e.g., ESRD) and some patients develop ESRD independent of their LN diagnoses (e.g., medullary sponge kidney disease, reflux nephropathy, and diabetic nephropathy), for a sensitivity analysis, we also completed extensive chart review to assess ESRD secondary to lupus nephritis (LN-related ESRD); LN-related ESRD was defined as dependence on dialysis (peritoneal and/or hemodialysis) and/or kidney transplantation in a patient with a history of LN.
Genotyping APOL1 and ancestry: Genotyping was performed using the Illumina Infinium® Expanded Multi-Ethnic Genotyping Array (MEGAEX) by the Vanderbilt Technologies for Advance Genomics (VANTAGE) core. Quality control was completed in PLINK version 1.90β3 following standard procedures, which include reconciling strand flips, removal of single nucleotide polymorphism (SNPs) with a call rate <0.95, and removal of samples with: (a) per-individual call rate <0.95; (b) inconsistent sex with the EHR; (c) duplicated pairs (PI-HAT ≥0.95); (d) relatedness (proportion identity by descent PI_HAT ≥0.25); and (e) compromised DNA; SNPs that departed from Hardy Weinberg Equilibrium (HWE) were also removed. 33 Principal components (PCs) were calculated with the SNPRelate package 34 and used in conjunction with HapMap populations 35 to define ancestry by including any subject within ±4 SD of the mean values for populations of recent European and African ancestry. Genetic data was imputed using the Michigan Imputation Server (HRC v1.1). 36
One of the G1 variants (rs73885319) was directly genotyped, while the other (rs60910145) was imputed (INFO R2 = 0.99). To define the G2 variant, we used rs12106505, an imputed variant (INFO R2 = 0.94) which is in complete LD with rs71785313 in individuals of AA in the US. 19 APOL1 HR genotypes were defined as G1/G1, G2/G2, or G1/G2; low risk (LR) genotypes were defined by the presence of 1 or 0 copies of the G1 and G2 alleles.
Patient Characteristics and Covariates: We collected information regarding sex, median age in the EHR, year of birth, age at SLE diagnosis (defined by the first phecode for SLE), and length of follow-up in the EHR for adjustment in statistical analyses. In addition, all serum creatinine levels, systolic (SBP) and diastolic (DBP) blood pressures recorded in the EHR were extracted. The highest serum creatinine was used as a measure of worst renal function, and the median values of SBP and DBP were calculated. The following blood pressure values were deemed to be questionable and excluded from the median calculation: SBP < 60 or >250 mm Hg, and DBP < 40 or >140 mm Hg. 37
Statistical analysis: Patient characteristics, including highest serum creatinine, are presented as number (percent) for categorical variables and median [IQR] for continuous variables. Pearson’s chi-square and Wilcoxon’s rank sum tests were used to assess categorical and continuous variables, respectively. Post-hoc detectable differences were calculated. For example, the allele frequency for the APOL1 HR genotypes was 18.3%, there were 40 cases of phecode-defined ESRD, and 85 controls among patients of AA in this cohort. Thus, we had 80% power to detect an OR ≥7.0 at a type 1 error of 0.05 in a recessive model.
We first performed a PheWAS comparing SLE patients of AA and EA to determine the clinical diagnoses related to renal and hypertensive disease that differed among SLE by ancestry. Then, to assess whether the observed significant associations were primarily attributable to APOL1 HR genotypes, we completed logistic regression analyses that excluded SLE patients with APOL1 HR genotypes, using ancestry as exposure and the renal and hypertensive-related diagnoses that differed significantly among patients of EA and AA in the preceding PheWAS as the outcomes. The prevalence of these diagnoses in AA patients with and without an APOL1 HR genotype among patients with SLE was not evaluated since power was limited. As a sensitivity analysis, we assessed the prevalence of LN-related ESRD using logistic regression.
All analyses were adjusted for sex, median age in the EHR, age at SLE diagnosis, and length of follow-up in the EHR. In the PheWAS, there were 362 clinical diagnoses with at least 20 cases (defined as ≥ 2 phecodes on separate days during follow-up) in our study population, yielding a Bonferroni-adjusted significant p-value ≤1.38E-4 (0.05/362). For the primary logistic regression analysis, a Bonferroni-adjusted p-value ≤2.78E-3 (0.05/18, where 18 was the number of selected renal and hypertensive-related clinical diagnoses that yielded significant associations in the PheWAS) was considered significant. For the sensitivity analysis, a p-value <.05 was considered significant.
PheWAS analysis was performed using the PheWAS R package 38 and the logistic regression with R software version 4.1.1. Associations are shown as odds ratio [95% confidence interval] (OR [95% CI]).
Results
Study population
Characteristics of the study population by ancestry.
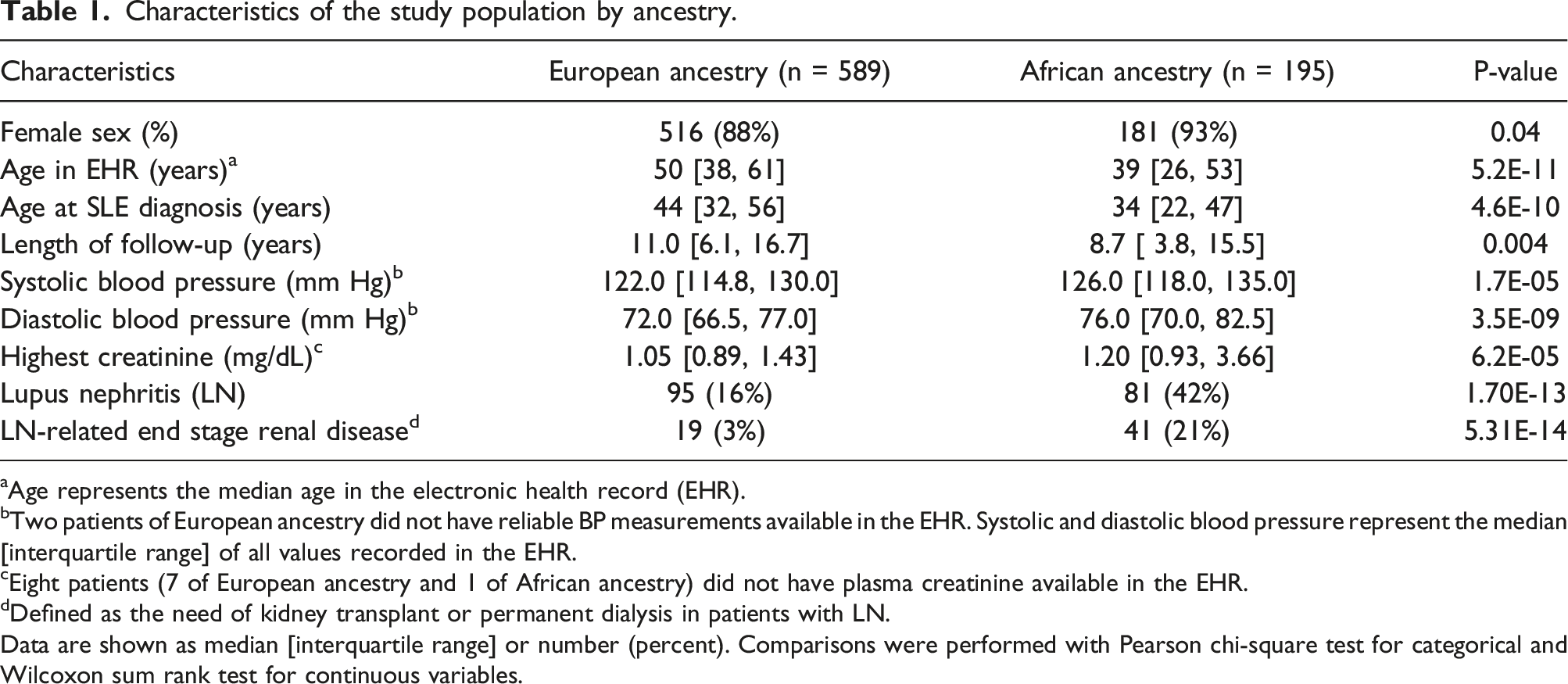
aAge represents the median age in the electronic health record (EHR).
bTwo patients of European ancestry did not have reliable BP measurements available in the EHR. Systolic and diastolic blood pressure represent the median [interquartile range] of all values recorded in the EHR.
cEight patients (7 of European ancestry and 1 of African ancestry) did not have plasma creatinine available in the EHR.
dDefined as the need of kidney transplant or permanent dialysis in patients with LN.
Data are shown as median [interquartile range] or number (percent). Comparisons were performed with Pearson chi-square test for categorical and Wilcoxon sum rank test for continuous variables.
APOL1 genotype
Characteristics of the study population by APOL1 genotype in patients of African ancestry with systemic lupus erythematosus.
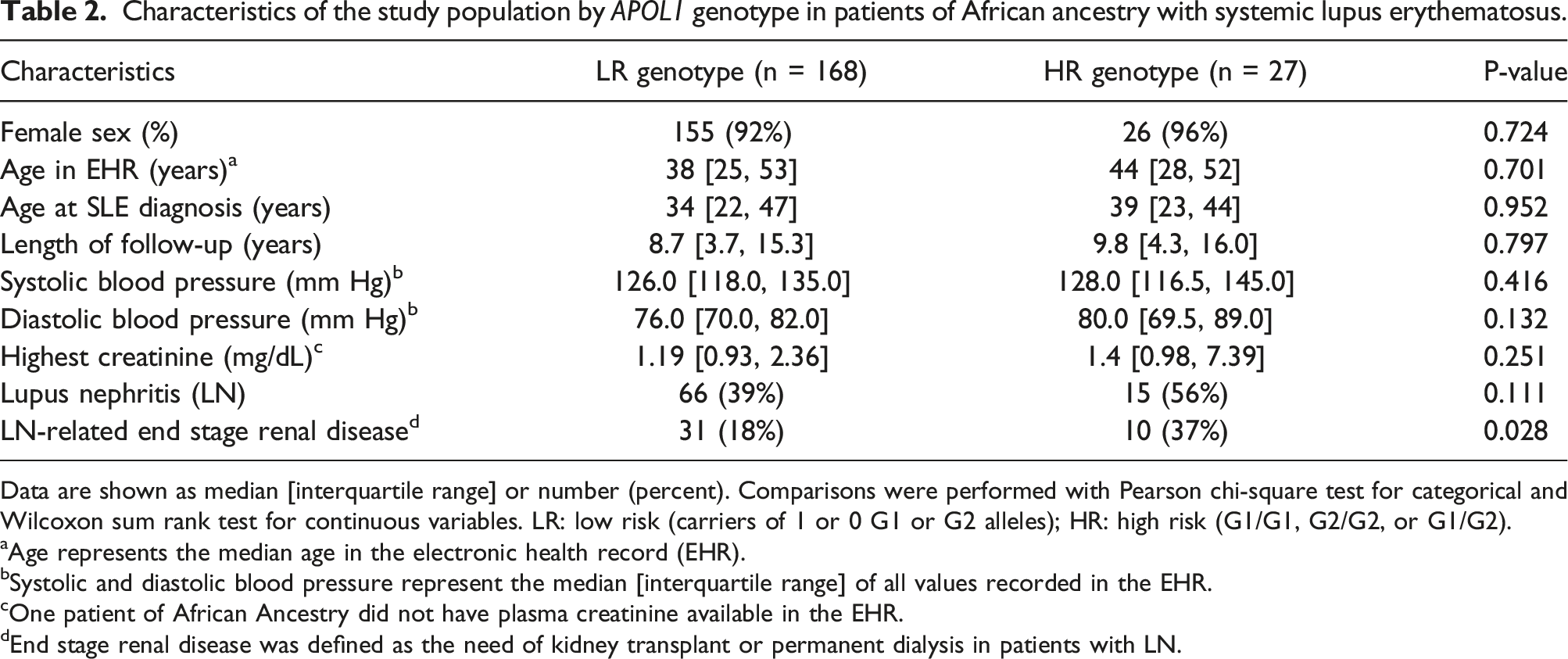
Data are shown as median [interquartile range] or number (percent). Comparisons were performed with Pearson chi-square test for categorical and Wilcoxon sum rank test for continuous variables. LR: low risk (carriers of 1 or 0 G1 or G2 alleles); HR: high risk (G1/G1, G2/G2, or G1/G2).
aAge represents the median age in the electronic health record (EHR).
bSystolic and diastolic blood pressure represent the median [interquartile range] of all values recorded in the EHR.
cOne patient of African Ancestry did not have plasma creatinine available in the EHR.
dEnd stage renal disease was defined as the need of kidney transplant or permanent dialysis in patients with LN.
Association between ancestry and APOL1 HR genotype with disease prevalence
African ancestry in SLE patients was associated with increased prevalence of 24 phenotypes compared with SLE patients of EA (OR≥2.6, p-value < 1.38E-04, Supplementary Table S1). Eighteen of these 24 phenotypes were related to renal and hypertensive disorders (Figure 1). The remaining six phenotypes were cardiomegaly, pleurisy/pleural effusion, anemia of chronic disease, pulmonary collapse/compensatory emphysema, congestive heart failure not otherwise specified, and pruritus and related conditions. Association between kidney-related disorders in patients with systemic lupus erythematosus and African ancestry. Y axes shows the selected phenotype and X axes shows the Odds Ratio and 95% confidence interval (95%CI) for each selected phenotype when comparing patients of African ancestry with European ancestry including individuals with APOL1 HR genotype (black dots and lines) and after excluding individuals with APOL1 HR genotype (red dots and lines). NOS: no other specified.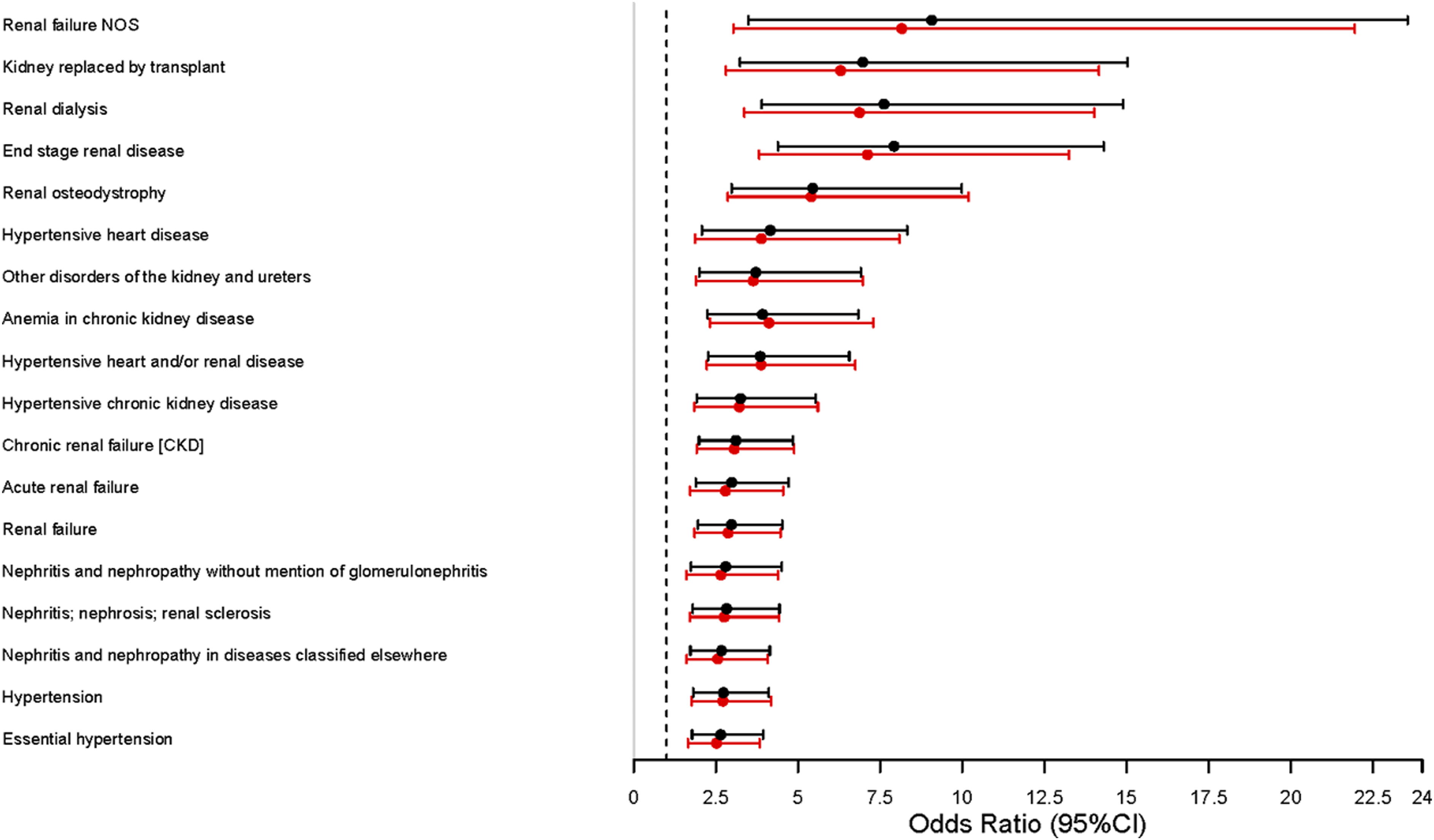
Logistic regression analyses for the 24 phenotypes of interest comparing SLE patients of AA with EA without individuals who carried an APOL1 HR genotype showed a small decrease in the odd ratios (Supplementary Table S2), but all associations remained significant (p-value <2.87E-3). All renal and hypertensive phenotypes that differed significantly in prevalence among EA and AA patients remained significantly different after exclusion of AA patients with HR APOL1 genotypes (Figure 1, Supplementary Table S2).
Sensitivity analysis for LN-related ESRD
SLE patients of AA showed an increased risk of LN-related ESRD compared to EA patients (OR = 9.02 [4.96, 17.0], p-value = 7.27E-12) after adjusting for sex, median age in the EHR, age at SLE diagnosis, and length of follow-up in the EHR in logistic regression analysis. Among SLE patients of AA, the APOL1 HR genotype was associated with increased risk of LN-related ESRD (OR = 2.56 [1.03, 6.17, p-value = 0.038) after the same adjustments; however, in a post-hoc exploratory analysis, the association was attenuated when PCs were included among the adjustments (OR = 2.23 [0.86, 5.56], p-value = 0.091).
Discussion
Our primary findings indicate that patients with SLE of AA have higher prevalence of several disorders compared to patients of EA, particularly those related to hypertension and kidney disease; however, the APOL1 high risk variants do not appear to fully explain these differences. The results are of particular importance given the stakes for identifying drivers of disparities among patients of AA and EA diagnosed with SLE. Not only does SLE occur more frequently in individuals of AA compared to EA, 40 but patients of AA experience earlier disease onset, more organ damage, and higher mortality rates. 41 Furthermore, patients of AA have higher rates of LN, neurological, and hematological manifestations than EA, 42 and they exhibit different therapy responses compared to EA patients.43–45 As such, therapies targeting the causes of these differences could help mitigate disparities; however, it is critical to ensure that these therapy targets are associated with disease-specific difference. This finding that patients of AA, regardless of APOL1 HR variants, experienced negative renal and hypertensive outcomes compared to patients of EA suggests that therapies targeting APOL1 likely will not ameliorate most of the observed disparities by ancestry in patients with SLE.
These results were consistent with previous findings that patients of AA experience higher risks of renal and hypertensive outcomes compared to patients of EA. Moreover, in our study, the APOL1 HR variants were present in 14% of the SLE patients of AA and in 19% of those with LN; this prevalence is similar to that reported in LN patients of AA who underwent kidney biopsy (17%). 46
While our numbers were too small to assess the risks for most hypertensive and renal outcomes among patients of AA alone based on APOL1 genotype status, we were able to assess LN-related ESRD in a sensitivity analysis. Given that ESRD has significant associations with each LN and APOL1 HR genotypes, it was important to carefully assess whether LN-related ESRD was associated with APOL1 HR genotypes among this cohort. Defining relationships and causality between complex conditions can be challenging using standard bioinformatics tools, and the fidelity of phenotype definitions using phecodes varies by phenotype. As such, we used extensive chart review to define a select group of patients with LN-related ESRD.
We observed that not only did AA patients experience higher risk of LN-related ESRD compared to EA patients, but also that the prevalence was higher for those with APOL1 HR genotypes compared to LR among patients of AA. A post-hoc analysis showed that this association was attenuated after adjustment for PCs, but cohort size limited the statistically supported degrees of freedom necessary to rely upon these additional adjustments.
In agreement with our finding, studies in SLE and in the general population suggest that APOL1 HR genotypes are only associated with severe forms, but not mild forms, of kidney disease, raising the possibility of a “double-hit” impact without independent effect. 47 Further, previous research among patients with SLE has found that while carriers of APOL1 HR genotypes have increased risk of developing collapsing nephropathy 46 and LN-associated ESRD, 12 not all carriers develop LN-associated ESRD. 12 Moreover, in vitro studies suggest that interaction with environmental factors that activate the innate immune system are also important, because APOL1 transcription is enhanced by inflammatory factors through both interferon dependent and independent pathways. 21
Regardless, these results demonstrate that even if the APOL1 HR genotype contributes to increased risk for selected renal and hypertensive outcomes, as found in prior research, 12 the APOL1 HR genotype is not the predominant driver of differences in outcomes between AA and EA patients with SLE, except for LN-related ESRD. Indeed, since risk by ancestry remained significantly elevated in these phenotypes when patients with the APOL1 HR genotype were omitted, other factors (e.g., genetic, social, or environmental) appear to play a larger role in racial discrepancies. Given that the majority of AA patients do not carry the APOL1 HR genotype and that the risk remains elevated by ancestry after excluding these individuals, treatments targeting APOL1 may not mitigate the majority of racial disparities in outcomes for patients with SLE.
This study has limitations: (a) while socioeconomic status was not included in our analysis, previous studies have shown that genetic ancestry strongly correlates with socioeconomic status, 48 and ethnic disparities in renal involvement seems to be explained better by ancestry than socioeconomic status;49,50 (b) the findings may not be generalizable to all patients but rather to those who received care at a tertiary care hospital; (c) there is potential misclassification bias since the quality of case-control definition varies across phenotypes when using billing codes to assemble phenotypes; however, our results are consistent with previous literature where SLE individuals of AA have higher prevalence of several renal and hypertensive related disorders; 41 (d) as noted above, our cohort was too small to detect most differences among patients of AA and limited the interpretation of LN-related ESRD by APOL1 status among AA patients; however, we had sufficient power to detect differences between individuals of AA and EA.
In conclusion, among patients with SLE, the higher prevalence of renal and hypertensive disorders in those of AA compared to EA are not primarily explained by the presence of APOL1 high risk variants in patients of AA.
Supplemental Material
Supplemental Material - APOL1 and the risk of adverse renal outcomes in patients of African ancestry with systemic lupus erythematosus
Supplemental Material for APOL1 and the risk of adverse renal outcomes in patients of African ancestry with systemic lupus erythematosus by Cecilia P Chung, Gul Karakoc, Alyson Dickson, Ge Liu, Jorge L Gamboa, Jonathan D Mosley, Nancy J Cox, and Vivian K Kawai in Lupus
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The study was supported by NIH/NIAMS grant R01AR076516 and the Lupus Research Alliance – BMS Accelerator Award. The dataset(s) used for the analyses described were obtained from Vanderbilt University Medical Center’s BioVU which is supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH funded Shared Instrumentation Grant S10RR025141; and CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at ![]() . CPC is funded by NIH/NIAMS R01AR073764, and JDM by NIH/NIGMS R01GM130791.
. CPC is funded by NIH/NIAMS R01AR073764, and JDM by NIH/NIGMS R01GM130791.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.