
Abstract
The association of dysregulated metabolism in systemic lupus erythematosus (SLE) pathogenesis has prompted investigations into metabolic rewiring and the involvement of mitochondrial metabolism as a driver of disease through NLRP3 inflammasome activation, disruption of mitochondrial DNA maintenance, and pro-inflammatory cytokine release. The use of Agilent Seahorse Technology to gain functional in situ metabolic insights of selected cell types from SLE patients has identified key parameters that are dysregulated during disease. Mitochondrial functional assessments specifically can detect dysfunction through oxygen consumption rate (OCR), spare respiratory capacity, and maximal respiration measurements, which, when coupled with disease activity scores could show potential as markers of disease activity. CD4+ and CD8 + T cells have been assessed in this way and show that oxygen consumption rate, spare respiratory capacity, and maximal respiration are blunted in CD8 + T cells, with results not being as clear cut in CD4 + T cells. Additionally, glutamine, processed by mitochondrial substrate level phosphorylation is emerging as a key role player in the expansion and differentiation of Th1, Th17, ϒδ T cells, and plasmablasts. The role that circulating leukocytes play in acting as bioenergetic biomarkers of diseases such as diabetes suggests that this may also be a tool to detect preclinical SLE. Therefore, the metabolic characterization of immune cell subsets and the collection of metabolic data during interventions is also essential. The delineation of the metabolic tuning of immune cells in this way could lead to novel strategies in treating metabolically demanding processes characteristic of autoimmune diseases such as SLE.
Keywords
Introduction
The link between metabolism and immunity has prompted the exploration of how dysregulated metabolism drives aberrant immune responses, and how this contributes to systemic lupus erythematosus (SLE) pathogenesis. Metabolomic and transcriptomic studies have provided corroborating evidence that several metabolic pathways and metabolites are enriched in peripheral immune cells of patients with SLE.1,2 However, the value of bioenergetic analysis and adenosine triphosphate (ATP)-contributing anabolic pathways in clinical metabolic analysis is still in the early stages, as is the role that type I interferons (IFNs) contribute to metabolic dysregulation (Figure 1). The emerging role of aberrant mitochondrial function and bioenergetics in the progression to SLE disease. The risk of progressing to SLE increases with the presence of inflammatory cytokines, autoantibodies and interferon signaling gene (ISG) signatures. Numerous factors, including genetic make-up, environment, diet, and stress, can also modify disease course and severity. Currently, the role of bioenergetic dysfunction is not as well-established in terms of whether it contributes to or occurs with an increased risk of SLE, nor is it clear whether specific immune cell subsets play prominent roles in contributing to risk. Additional questions remain as to whether these cells show changes in their oxygen consumption rates that are measurable and detectible in preclinical disease and after formal diagnosis. Improved classification systems for preclinical SLE are needed to allow tailored early intervention and treatment strategies. Thus, circulating leucocytes or lymphocytes may act as early sensors or predictive biomarkers of mitochondrial function under conditions of systemic inflammation, having both diagnostic and prognostic value. Further, since the finding that glutaminolysis is altered in various T and B immune cell subsets in SLE, it still remains unknown whether glutamate oxidation contributes to inflammation and what its role might be in driving the progression of SLE.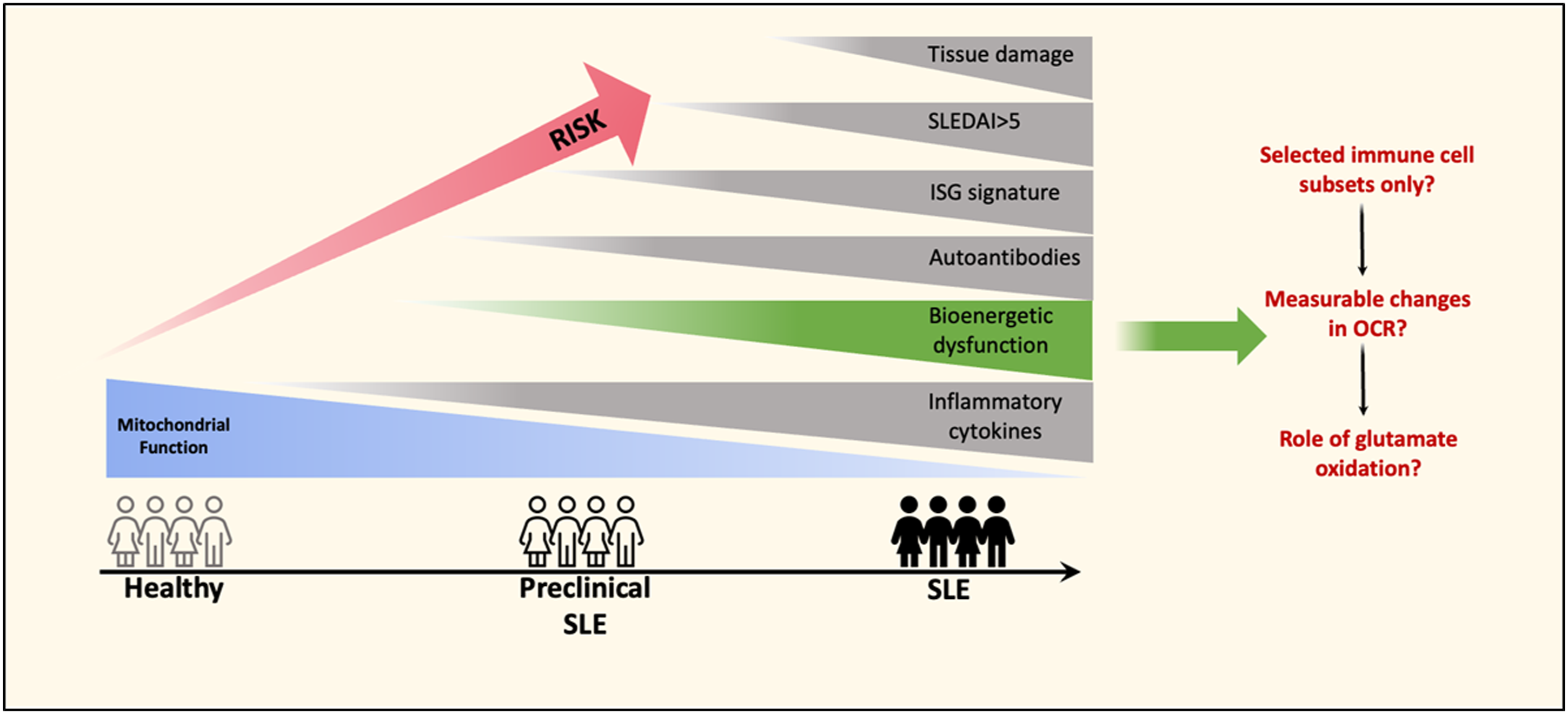
Immunological activation in response to invading pathogens or self-antigens results in a rapid upregulation in the metabolic response in immune cells of the innate and adaptive immune systems, causing them to proliferate and initiate their effector functions. These processes require not only an increased uptake of nutrients (mostly glucose, fatty acids, and glutamine) but also a switch to specialized metabolic pathways that correspond to specific immune effector functions. These processes require a significant supply of energy in the form of ATP which is produced via mitochondrial respiration oxidative phosphorylation (OXPHOS) or the breakdown of glucose (glycolysis). Metabolic processes of immune cells are distinct in comparison with somatic cells and can also differ substantially between immune cell subsets. Therefore, when determining metabolic therapies for autoimmune diseases such as SLE, individually assessing immune cell metabolism could be beneficial. Immunomodulating therapies are essential in SLE for the alleviation of symptoms and for the timely control of inflammation which reduces organ damage over time. The effects of immunosuppressives on metabolic parameters of SLE immune cells are surprising as they have little to no effect. This was demonstrated in CD4+ T cells that showed that the addition of dexamethasone, hydroxychloroquine, and mycophenolate mofetil for 24 h before Seahorse analysis(a real-time measure of cellular OXPHOS and glycolysis) had no effect in reducing OXPHOS and glycolysis in the SLE CD4+ T cells, 3 with both parameters remaining above control baseline measurements. This is a noteworthy finding and suggests that steroidal treatment may have a symptomatic rather than a therapeutic effect, or, that CD4+ T cells do not metabolically respond to immunosuppressive treatment. Either way, this points to aberrant metabolism as a prominent role player in SLE. The molecular complexity of autoimmune diseases such as SLE makes targeting specific cytokines challenging, therefore, guiding research endeavors toward evolutionarily conserved bioenergetic and biosynthetic pathways in immune cell subtypes could circumvent the molecular intricacies of autoimmune diseases, thereby improving or redefining treatment approaches.
This review aims to address recent, albeit few studies in SLE that have utilized in situ bioenergetic analysis by Seahorse respirometry to metabolically characterize human immune cells from SLE patients and the importance of these findings in disease pathogenesis and treatment.
Immune cell subsets from SLE patients exhibit altered functional metabolic parameters
Spontaneous and sustained type I interferon (IFN) induction is a hallmark and causal factor in SLE and other autoimmune diseases and is shown to be associated with metabolic and bioenergetic changes in several peripheral immune cell subsets. 4 During disease, the inflammatory environment induced by type I IFN results in metabolic rewiring of immune cells; 5 however, these effects are not consistent across subsets, with some cells showing more pronounced metabolic changes than others. Indeed, it has also been shown that defective mitochondrial clearance in non-immune cells (such as erythrocytes) can indirectly cause metabolic rewiring in macrophages leading to aberrant type I IFN signatures in SLE. 6
Naïve T cells or resting T cells have a low energy demand and utilize OXPHOS to generate ATP during immune surveillance, however, activated T cells show an increase in glycolysis and mitochondrial metabolism to meet the biosynthetic demands of the cell. In SLE patients, metabolic changes have primarily been reported in CD4+ and CD8+ T cells which have shown amplified energy metabolism and increased mitochondrial transmembrane potential, suggesting that even at a resting state these immune cells are primed for a rapid upregulation in effector function. 4 Autoreactive CD4+ T cells play an essential role in SLE pathogenesis by associating with autoantibody-producing B cells. It was noted that these CD4+ T cells had increased OCR, ECAR, and spare respiratory capacity in comparison to CD4+ T cells from healthy controls with and without anti-CD3 and anti-CD28 activation. 3 Normally, SLE patients present with a reduced frequency of naïve CD4+ CD45RA + CCR7+ T cells (Tn) together with a concomitant increase in CD4 + CD45RA−CCR7+ central memory T cells (Tcm), which alters the ratio of Tn/Tcm subsets. The same study found that the percentage of Tn was inversely correlated with both activated ECAR and basal OCR levels and that Treg percentages were positively correlated with activated ECAR, implicating metabolic dysfunction of these cells in SLE disease. Recently, the analysis of CD4+ T cells from SLE patients was reassessed similarly, however, no significant changes in basal and maximal OCR, as well as spare respiratory capacity, were observed. 4 More functional studies on CD4+ T cells from SLE patients are required to confirm whether metabolic dysregulation is a feature of these cells in SLE.
CD8+ T cells in SLE have dampened cytotoxic functions that can lead to an increased risk of infection, which is the highest cause of death in SLE. 7 A study comparing the metabolic activity of CD8+ T cells from SLE patients with no expression of IFN signaling genes (ISG) to those with high ISG found that lower basal and maximal OCR, as well as spare respiratory capacity, was observed in CD8 + T cells from the high ISG cohort. This finding is significant since mitochondrial spare respiratory capacity is required for the cell to adapt to increases in energy demands, such as during an infection. Research has shown that normal mitochondrial respiration restricts the growth of the bacterial pathogen Listeria monocytogenes. 8 Thus, healthy mitochondria likely drive adaptive immune responses in CD8+ T cells. Interestingly, glycolysis, measured by ECAR showed no significant differences between healthy controls and SLE patients, 4 indicating that the association of glycolysis and pathogenesis in CD8+ T cells is unlikely.
Among the innate effectors, monocytes are specialized immune cells that show remarkable metabolic flexibility. They can sequentially mount anabolic energy-consuming immune activation events, switch to catabolic energy-conserving processes during immune deactivation, and re-establish anabolic and catabolic energy balance to restore homeostasis. 9 They are poised to be rapidly mobilized in large numbers to inflamed sites throughout the body, where they serve to provide pro-inflammatory or resolving activities. During SLE and rheumatoid arthritis (RA), monocytes have a distinct role in pathogenesis by inducing robust pro-inflammatory features. 10 It was observed that SLE monocytes exhibit an increased glucose-dose dependent glycolytic flux which can be phenocopied by treating healthy monocytes with IFNα. 11 Similarly, in individuals at risk of developing RA, it was observed that metabolic priming was detected in CD14+ monocytes, which was a phenotype that preceded the clinical manifestation of the disease. Additionally, baseline OCR was increased in these monocytes and exhibited a glycolysis-driven inflammatory phenotype. 12 Interest in monocyte subsets aside from the CD14+ CD16-classical monocytes, and their involvement in SLE pathogenesis is gaining traction; 10 however, an in-depth assessment of their metabolism and correlation with SLE disease activity is yet to be determined.
Mitochondrial metabolism is altered in SLE
Mitochondrial dynamics and metabolism are interlinked to drive immune cell function and fate. This relation provides an exciting model by which mitochondrial dynamics can be used as a nodal point to integrate and shape immunometabolism and function. 13 Mitochondrial nucleic acid-induced type I IFN secretion has been found to drive autoinflammation as a result of defective mitophagy systems 14 causing the accumulation of dysfunctional mitochondria. Similarly, mitochondrial abnormalities can be phenocopied by exposing various immune cells from healthy volunteers to type I IFN in vitro.4,11 Mitochondrial functional assessments can detect dysfunction through OCR, spare respiratory capacity, and maximal respiration measurements which have the potential to act as a biomarker of disease activity 15 which in theory could be applied to SLE. These assessments have also been used to assess the therapeutic potential of drugs in treating SLE through the regulation of mitochondrial function. For example, the use of idebenone, a synthethic Coenzyme Q 10 analog and antioxidant, has been shown to improve mitochondrial metabolism (30% increase in basal respiration and ATP production), reduce the expression of type I IFNs, and ameliorate murine lupus disease activity in lupus-prone mice. 16
The fact that type I IFNs have been shown to affect mitochondrial functions in many peripheral immune cell subsets links with previous evidence supporting an emerging concept that circulating leucocytes act as early sensors or predictive biomarkers under several disease conditions. 15 The impact of these dysfunctional mitochondria on functional bioenergetic parameters has not yet been defined in all immune subsets in SLE, however, some recent work has shed light on mitochondrial functional capacity before and during disease.
Nicotinamide adenine dinucleotide (NAD+) acts as a cofactor in multiple reactions related to energy production including the regulation of mitochondrial functional capacity. To regenerate mitochondrial membrane potential, NAD+ and FAD are converted by tricarboxylic acid (TCA) cycle enzymes back to their reduced forms. Interestingly, during SLE disease NAD+ is depleted in monocyte and CD8+ T cell populations, which has recently been discovered as being due to CD38, an enzyme expressed during SLE that degrades NAD+. CD38 expression and presumably NAD+ depletion were shown to translate into decreased basal OCR and maximal respiration in CD8 + T cells from SLE patients. 17 These results were corroborated by another study that showed that IFNα exposure increases NAD + consumption in CD8+ T cells through upregulation of NAD-consuming enzymes and pathways 4 and agreed with functional assessments showing that CD8+ T cells exhibited decreased spare respiratory capacity in comparison to healthy controls. 4
The presence of NAD+ and its role in maintaining normal metabolism was presented in a recent study that found that both NAD+ production and consumption in monocytes are increased in inactive or early SLE disease, potentially as a component of intracellular monocytic anti-inflammatory programming. Lower cellular NAD+ levels in monocytes were found to reduce the inflammatory response to LPS by inhibiting TLR4 signaling, 18 indicating the requirement of NAD+ for the resolution of inflammation. This was confirmed by a study that found that the addition of the NAD+ precursor nicotinamide riboside (NR) increased NAD+ levels and attenuated the type I IFN signaling pathway in monocytes. 19 NAD+ metabolism may therefore be a promising target pathway to rebalance OXPHOS in cells that are crucially involved in SLE pathogenesis.
Finally, a recent study has shown that mitochondria from SLE B cells are hyperpolarised which significantly correlated with SLEDAI scores 20 and that enhanced mitochondrial functions mediated by mitochondrial fuel switching during hyperpolarisation are important for plasmablast differentiation. 20
Aberrant OXPHOS drives an inflammatory phenotype
In the resting phase, immune cells utilize OXPHOS as a source of energy production. As mentioned previously, metabolic reprogramming causes a switch to glycolysis in the advent of immune cell activation for fast turnaround of ATP, despite OXPHOS being the more energetically efficient process. Pro-inflammatory cytokines, chemokines, and metabolites are secreted during this state, thus linking glycolysis with the onset and sustenance of inflammation. As homeostasis returns, the energetic demands of immune cells reduce, and they revert to OXPHOS with the simultaneous release of anti-inflammatory cytokines.
However, an increasing number of studies show that this divide is not as clear cut and suggests a prominent role of OXPHOS in driving inflammation that is underpinned by reactive oxygen species (ROS) release. ROS are primarily produced by NADPH oxidases (NOX) and mitochondrial Electron Transport Chain (ETC) complexes I and III as a normal product of OXPHOS (mtROS). 21 They are important secondary signaling messengers, and their role in the maintenance of cellular oxidative homeostasis, propagation of signaling pathways, and crosstalk with other organelles through the formation of “microdomains” has been previously well-described. 22 Mitochondrial ROS are involved at different points in the inflammatory process, and even possibly as a regulator of inflammatory responses. 23 Oxidative stress in general has been shown to trigger the activation and assembly of the NLRP3 inflammasome, subsequently causing the release of pro-inflammatory cytokines such as IL-1β and IL-18, which can trigger innate immune defenses. 23 While NLRP3 activation can theoretically also be caused by ROS from non-mitochondrial sources such as NOX1, NOX2, and NOX4, several studies have dismissed these, therefore maintaining mtROS as the primary source for ROS-mediated-NLRP3 activation. 24
This OXPHOS-associated pro-inflammatory phenotype is heightened in COPD, critical illnesses, and SLE among other diseases, across various cell types.25,26 While mtROS production is beneficial to many cellular processes, aberrantly increased production of mtROS during chronic inflammation can begin to affect the mitochondria itself.
27
For example, in inflammatory macrophages, excess mtROS are both produced by mitochondria, as well as target them. This causes mtDNA leakage which then functions in the capacity of a DAMP (Figure 2). When released into the cytosol, mtDNA activates the NLRP3 inflammasome further enhancing the production of pro-inflammatory cytokines, and once secreted into extracellular plasma enhances systemic inflammation.
28
The overall inflammation, in turn, promotes mtROS accumulation causing the development of a vicious cycle centered around the continuance of mitochondrial dysfunction.
29
Further, such sustained production of mtROS, and thus sustenance of a pro-inflammatory environment, can affect intra-organelle crosstalk potentially leading to the development of autoimmunity.
22
Indeed, increased mtROS production along with persistent mitochondrial hyperpolarization, ATP depletion, and increased type I IFN responses have been reported in SLE,
30
together with enhanced IL-1β and IL-18 secretion, and this correlates with disease progression in mouse studies of SLE.
31
Combined with existing implications of dysregulated mitochondria, and therefore OXPHOS in SLE, this suggests that mtROS-mediated NLRP3 inflammasome activation might play a part in the autoimmune, autoinflammatory phenotype associated with this disease. Such effects have been noted in several immune cell types such as CD8+ T cells, low-density neutrophils, and plasmacytoid dendritic cells (pDCs) specifically in the context of SLE.4,32 Further, several human studies also back the potential involvement of the NLRP3 inflammasome in SLE in some form, whether as a risk factor, a susceptibility factor, or even as a cause of pathogenesis itself.
33
Mitochondrial ROS (mtROS)-mediated mitochondrial dysfunction causes an inflammatory phenotype through NLRP3 inflammasome activation, disrupted maintenance of mtDNA, and pro-inflammatory cytokine release. (A) When excess mtROS production exceeds its normally beneficial functionality in cellular processes, it causes negative offshoot effects. Such effects have been noted in autoimmune conditions such as SLE where immune cells develop an inflammatory phenotype mediated by excess mtROS production (by way of damaged OXPHOS). This excess mtROS in turn drives the assembly and activation of the NLRP3 inflammasome, which causes the release of pro-inflammatory cytokines IL-1β and IL-18. mtROS also causes disruption of mtDNA maintenance causing it to leak from mitochondria into the cytosol and extracellular spaces. This extruded mtDNA functions as a DAMP and drives the release of IFN𝛼, further sustaining the inflammatory phenotype through autocrine as well as paracrine effects. (B) A vicious cycle develops, leading to further mitochondrial dysfunction, eventually leading to persistence of mitochondrial hyperpolarisation, ATP depletion, and type I IFN responses.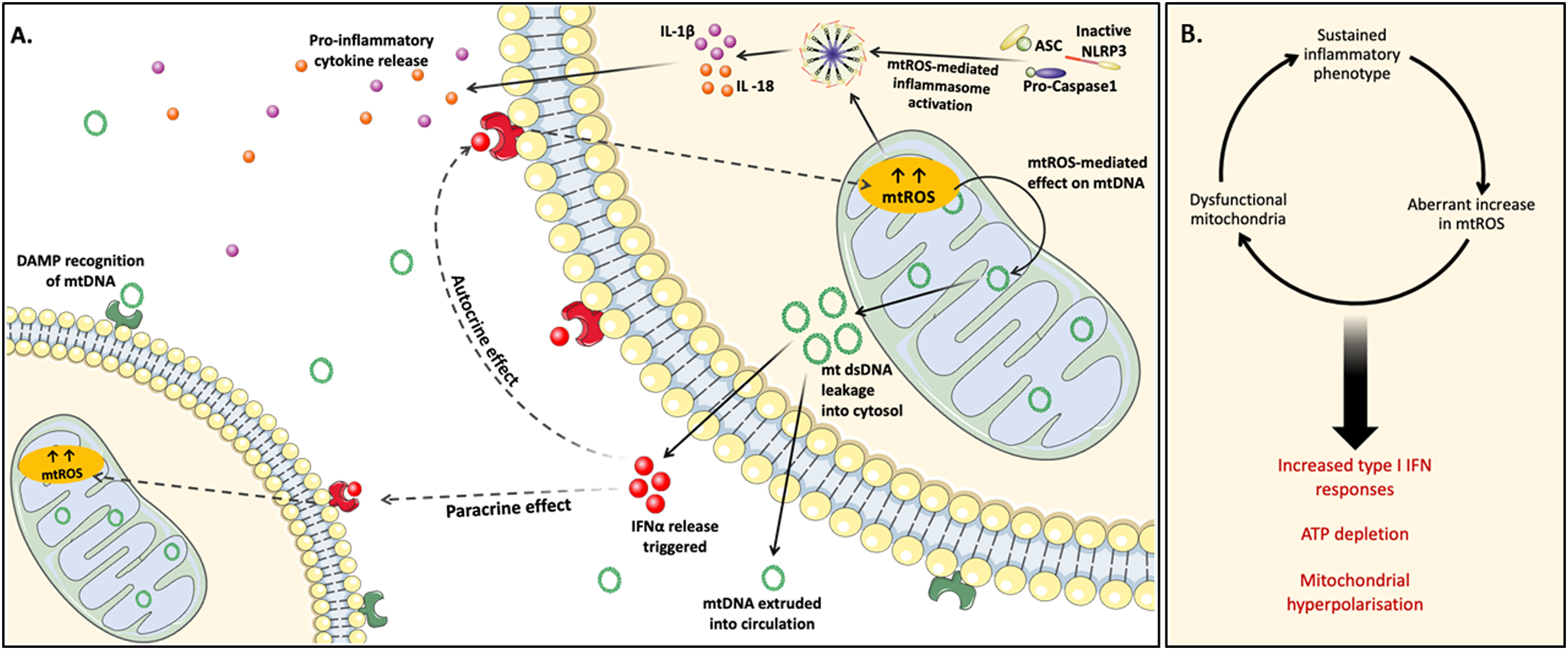
Altered OCR as a marker of disease activity?
Basal respiration can be considered the threshold below which the cell cannot sustain OXPHOS to meet energy demands 15 and is different between immune cell populations. In cancer, higher OCRs have been noted, and this more often than not co-exists with glycolytic compensation owing to defective and/or damaged mitochondria. It has been suggested that the identification of increased OXPHOS as a predictor of chemotherapy resistance could be possible. 34 To understand how this could be applied to SLE, a comprehensive bioenergetic analysis of the peripheral immune cell subsets will need to be conducted where correlations between OCR, spare respiratory capacity, and maximal respiration measurements are correlated with SLEDAI scores. In doing so, cell subtypes associated with metabolic aberrations can be pinpointed and further assessed in detail. Changes in bioenergetic function and OCR in patient immune cell populations can then reflect both metabolic stress and the changing role of these cells in immunity and inflammation.
Lymphocytes are a heterogeneous population of cells, which are normally in a quiescent state and are reliant on mitochondria to meet their energetic demands. Activated CD4+ T cells are a common feature of SLE and exhibit increased OXPHOS, making these cells an appropriate choice as an indicator of disease activity. Interestingly, mitochondrial respiration is required for ISG expression in CD4+ T cells 35 and points to increased OCR as a potential predictor of active disease/therapy failure, or conversely, a reduction in OCR could be applied as a predictor of treatment success. The inhibition of OXPHOS (measured through a decrease in OCR) by inhibitors of the ETC complexes I, III, and IV such as metformin successfully decreased CD4+ T cell activation in humans as well as in mouse models.3,35 The use of metformin and the concurrent decrease in CD4 + T cell activation may even be linked by the stage of activation at which it is administered. This might suggest the use of a combination of metabolic inhibitors applied at different stages of CD4 + T cell activation to produce significant therapeutic effects.3,35 Therefore, the link between increased OCR in CD4 + T cells and metabolic activation should be considered for its application in understanding treatment outcomes, as well that it could be expanded to other immune cell subsets that have not yet been assessed.
Mitochondrial substrate level phosphorylation as a marker of inflammation: A focus on glutamate oxidation
Glutamine is a conditionally essential, anaplerotic amino acid that is metabolized into glutamate and subsequently α-ketoglutarate which enters the TCA cycle. This process drives OXPHOS and generates two ATP molecules through succinyl-CoA synthetase in a reaction called mitochondrial substrate level phosphorylation (mSLP, Figure 3). This process generates substantial amounts of ATP in the absence of glucose or when mitochondrial integrity is compromised.
36
It is not surprising then that addiction to glutamine has been documented in almost all subtypes of cancer. Therefore, metabolic programming and the potential to stratify patients according to glutamine dependency is suggested to be an important determinant of responses to both conventional therapy and metabolic drugs in cancer treatment.
34
Emerging evidence showing increased dependency of B cells and T cells on glutamate oxidation suggests that this approach may be applied to SLE. The role of glutamine oxidation in B cell and T cell expansion and its possible role in driving pathogenesis in SLE. (A) Chronic type I IFN exposure induces metabolic rewiring in favor of glutaminolysis in B and T cells. Glutamine is converted to glutamate via glutaminase (GLS1) to drive TCA cycle intermediaries, a process known as mitochondrial substrate level phosphorylation (mSLP). This process generates substantial amounts of ATP in the absence of glucose or when mitochondrial integrity is compromised. It is an oxygen consuming phenotype that facilitates B cell differentiation to plasmablast cells, and T cell differentiation to Th17, Th1 and γδ T cells during active disease. (B) Does glutamate oxidation or dependency in B cells and T cells during active disease underpin pathogenesis and do current treatment regimes rebalance mitochondrial fuel usage?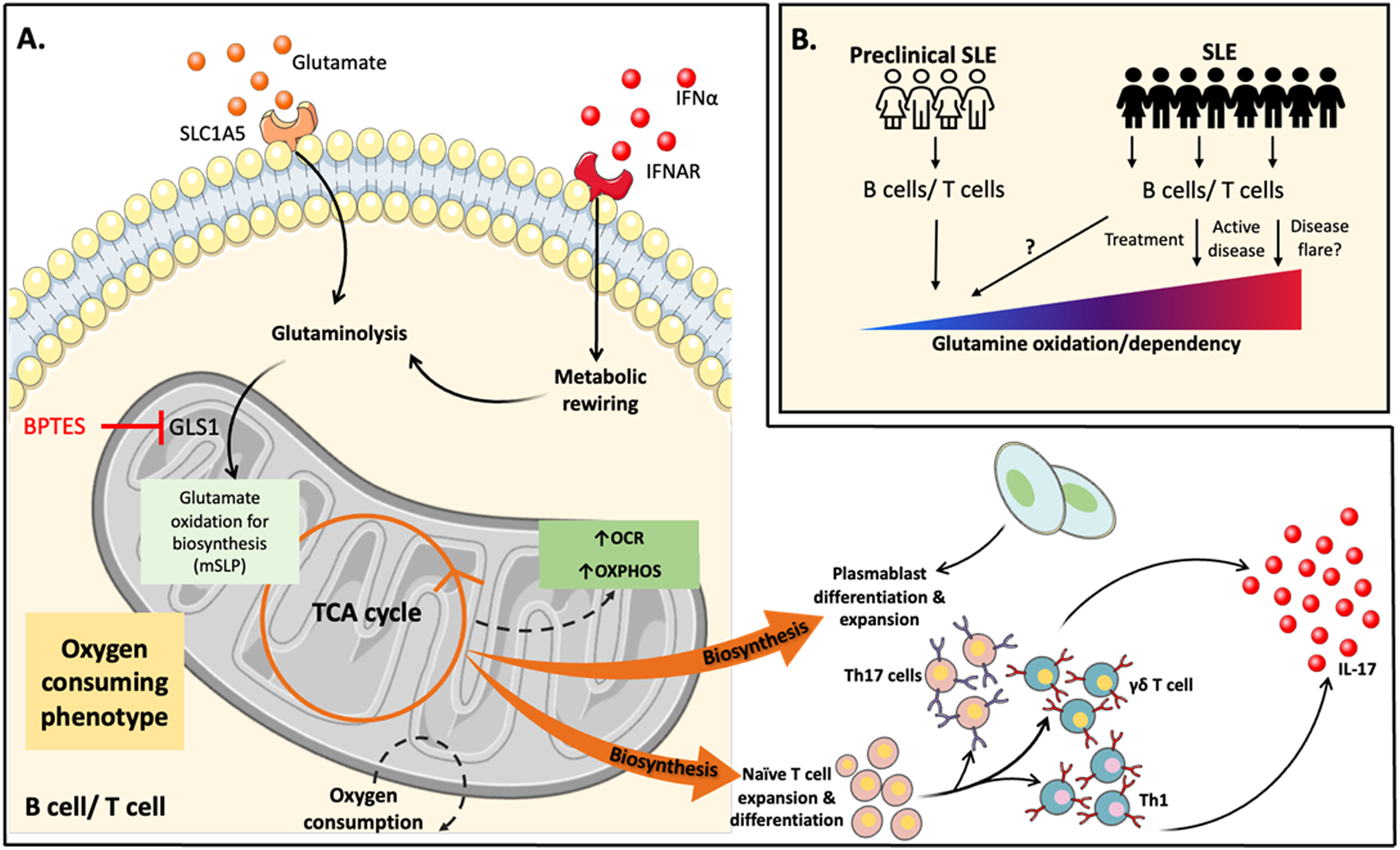
Recent research has shown that glutaminolysis is altered in various T cell immune cell subsets in SLE. 37 Glutamine was observed to be critically required for naive CD4 + T cell differentiation to Th1 and Th17 inflammatory T cells but not to Th2 or Treg cells. 38 Interestingly, previous studies in experimental mouse models of lupus have found that through specific inhibition of Glutaminase 1 (GLS-1, the first enzyme required for glutaminolysis), it is possible to downregulate glycolytic energy metabolism and reduce the differentiation of Th17 cells. 39 The role of glutamate oxidation in inflammation is also emerging– glutamine metabolism is essential for the production of IL-17A in γδ T cells during skin inflammation 40 and succinate synthesized via glutamine-dependent anaplerosis promotes the polarization of M1 macrophages. It is also noteworthy that glutamine catabolism can damage mitochondria due to ROS produced from an energized ETC and direct action of NH3 on Complexes I and III, 41 however, this hasn’t been confirmed in immune cells from SLE patients.
Mitochondrial dysfunction through enhanced glutamate oxidation in B cells from patients with SLE was recently shown to be involved in plasmablast differentiation and disease activity. Plasmablasts, defined as CD27hiCD38+, exhibit unique cellular properties compared with other B cell subsets (e.g., naïve and memory B cells) and plasma cells. In a recent study to investigate the role of glutamate oxidation in B cell differentiation and pathogenesis in SLE patients, it was observed that in the absence of glutamine, both glycolysis and OXPHOS were reduced, and plasmablast differentiation was suppressed, whereas there was no change in plasmablast differentiation in the absence of glucose. They also observed that by blocking the transport of glutamine into the mitochondria using BPTES, OXPHOS, ROS production, ATP production, and plasmablast differentiation decreased without affecting glycolysis. 20 Thus, in an inflammatory environment such as SLE, glutaminolysis may play a noteworthy role in pathogenesis (Figure 3). Future studies should interrogate these findings further before and after therapy.
A role for mitochondrial bioenergetics in SLE early disease detection
Mitochondria are both a source and a target for oxidative stress, therefore, the failure to maintain mitochondrial quality by the appropriate balance of biogenesis and autophagy leads to mitochondrial dysfunction. In SLE, mitochondrial dysfunction is very well characterized which is also observed in other chronic diseases such as diabetes. 43 These findings have formed the translational basis that circulating leukocytes can serve as bioenergetic biomarkers of systemic exposure to metabolic stressors or pro-inflammatory cytokines in diseases such as diabetes and cardiac surgery, 44 however, this is yet to be applied to SLE. Further, metabolomics and bioenergetic parameters derived from the Agilent Seahorse Cell Mito Stress test are well correlated with over one hundred metabolites, 45 supporting the accuracy of the Cell Mito Stress test in measuring bioenergetic health.
The early diagnosis of SLE is still an unmet need for many patients. 46 The preclinical lupus phase (preclinical SLE/pSLE) is defined as the time of an emerging maladaptive immune response before full-blown SLE. 47 Type I IFNs are overexpressed in preclinical SLE and are said to be directly implicated in the development of clinical autoimmunity. 48 Specifically, IFNα was found to be elevated in several cohorts before the development of SLE. 49 The available detection methods for pSLE lack sensitivity and improved classification systems are needed to allow tailored early intervention and treatment strategies. Circulating leucocytes or lymphocytes may therefore act as early sensors or predictive biomarkers of mitochondrial function under conditions of systemic inflammation. If bioenergetic health could be measured from these parameters at the “point of care,” it could have both diagnostic and prognostic value.
Therefore, from a precision medicine perspective, it becomes important to create diagnostic and prognostic bioenergetic health indices that may be applied to SLE.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Royal City of Dublin Hospital Trust.