
Abstract
Keywords
Introduction
Placental site trophoblastic tumor (PSTT) is a rare subtype of gestational trophoblastic disease with fewer than 300 cases reported in the literature. 1 It is usually diagnosed within days to months after labor, miscarriage or abortion, 2 but intervals of 10 years between pregnancy and diagnosis have been reported. 3 The association of PSTT and renal diseases is exceedingly rare with only 8 case reports published in which PSTT was found to be associated with nephrotic syndrome (Table 1). In those cases the renal biopsies showed one patient with membranous glomerulopathy, 4 six patients with thrombotic microangiopathy (TMA)5–9 and one patient with lupus nephritis. 10 In all cases hysterectomy was performed within months after onset of symptoms, leading to immediate remission of the nephrotic syndrome, except for one patient who died of complications of PSTT. 7
Cases presenting with lupus-like disease in relation to trophoblastic tumors.
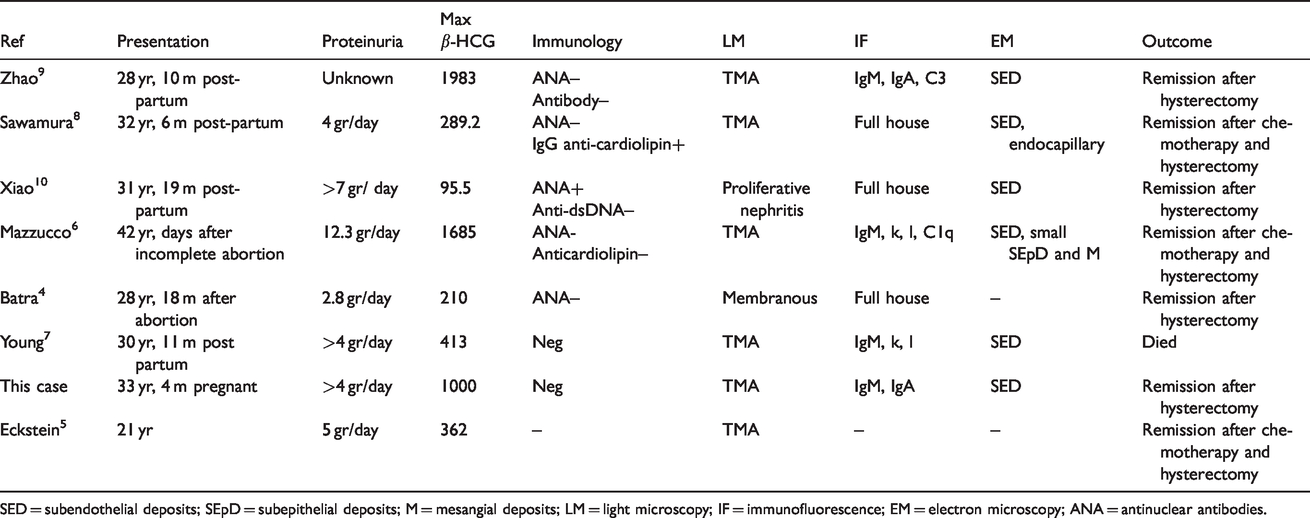
SED = subendothelial deposits; SEpD = subepithelial deposits; M = mesangial deposits; LM = light microscopy; IF = immunofluorescence; EM = electron microscopy; ANA = antinuclear antibodies.
We report the case of a patient with a lupus-like mesangiocapillary nephritis in which a clear connection between the discovery of PSTT and the nephrotic syndrome was found years later. During this delay the patient developed multiple lupus-like symptoms such as Raynaud’s phenomenon, protein losing entheropathy (PLE), and nodular regenerative hyperplasia (NRH). 11 A 'full' lupus-like syndrome such presented in this report, has to our knowledge never been described before in relation to a PSTT.
Case presentation
A 38-year old woman, gravida 2 para 2, presented to an affiliated hospital in June 2012 with hypertension (blood pressure 155/90 mmHg), generalized edema, and an erythematous rash across the cheeks and bridge of the nose. Additional testing revealed proteinuria of 5.3 g/24 h without erythrocyturia and with a normal kidney function (serum creatinine 55 µmol/L, eGFR 107 ml/min/1.73 m2, calculated using the MDRD formula).
Antinuclear antibodies (ANA) were absent and the levels of C3 and C4 were within normal range. The patient had not menstruated for over a year, after years of irregular blood loss following her last pregnancy 12 years prior to presentation. Therefore, she was referred to a gynaecologist. Surprisingly, the β-HCG level was 60 IU/mL (reference 0–1 IU/mL) but an abdominal ultrasound showed no abnormalities. A kidney biopsy was performed, which showed mesangiocapillary glomerulonephritis with extensive PAS+ pseudothrombi and pseudo-fullhouse immunofluorescent subendothelial and mesangial immune complexes. Based on light, immunofluorescence and electron microscopy there was a high suspicion of the presence of cryoglobulins (Figure 1). Serum cryoglobulins were tested three times over the disease course and were always absent. Upon suspicion of lupus nephritis, the patient was admitted for treatment with prednisone and mycofenolate mofetil (MMF) resulting in partial remission of proteinuria (hence 1.65 g/24h).
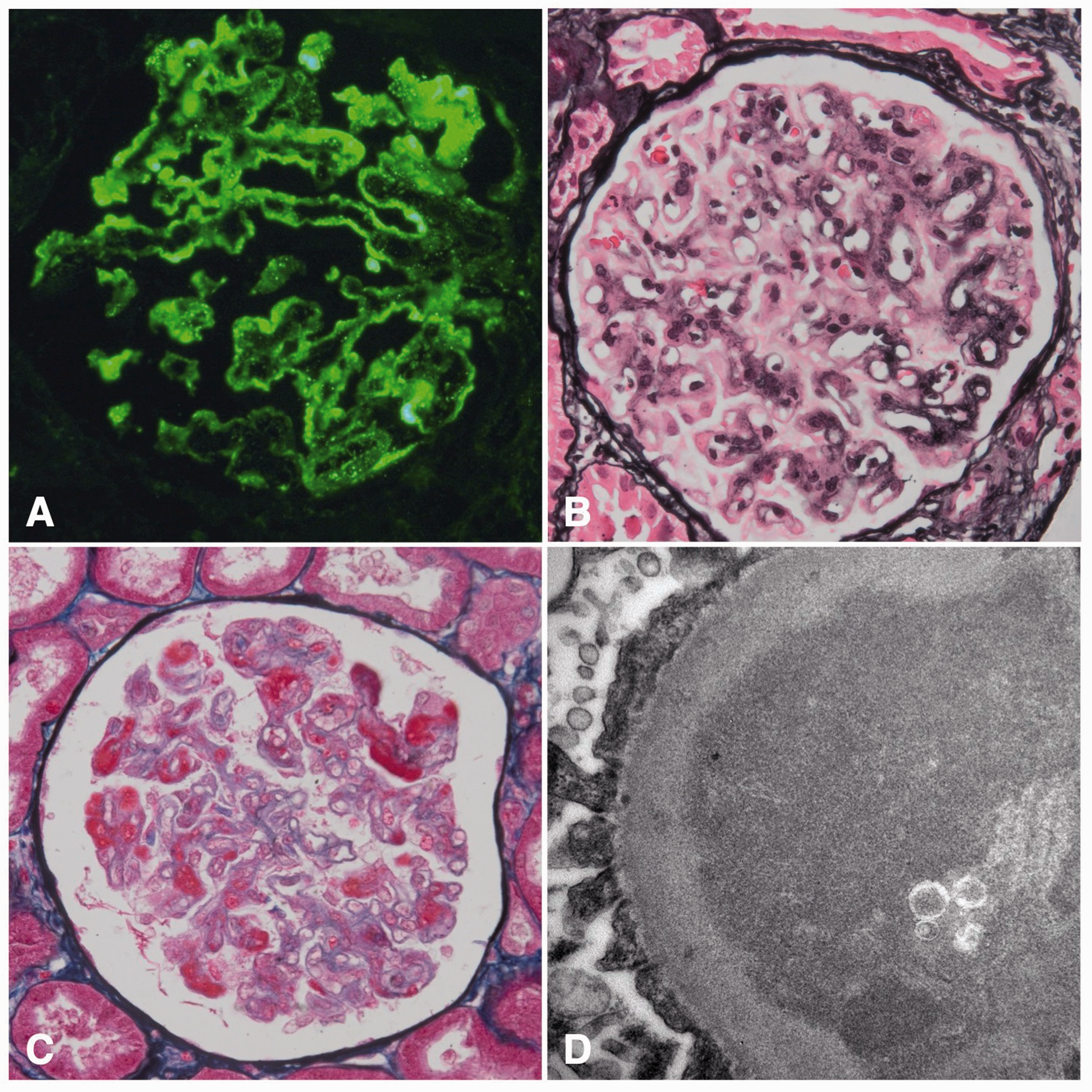
Immunofluorescence of IgM (A), light microscopy (B + C) and electron microscopy (D).
In October 2012, our patient reported abdominal discomfort which appeared to be secondary to ascites. Aspiration of ascites showed reactive fluid without signs of malignancy or infection. The nephrotic syndrome was still in remission (proteinuria of 0.8 g/24h), β-HCG had decreased to 36 IU/mL. The ascites progressed in the following years upon which the patient was referred to the Amsterdam University Medical Centers. Liver MRI and biopsy showed NRH with portal hypertension. A 99mTc human serum albumin scan and subsequent duodenoscopy with biopsy showed esophageal varices and a protein losing enteropathy (due to lymphangiectasia in the duodenum). The patient also had Raynaud’s phenomenon since the onset of the nephrotic syndrome in 2012; capillary microscopy revealed no morphological abnormalities. Despite the absence of ANA, the symptoms were explained as manifestations of systemic lupus erythematosus (SLE), because an alternative diagnosis was deemed unlikely.
Tumor markers were monitored and increased over time (β-HCG 172 IU/mL (Figure 2), CA-125 1955 IU/mL in August 2014). Ultrasound, CT, and MRI showed no abnormalities of the uterus or ovaries, besides uterus myomatosus, so watchful waiting was continued. Meanwhile, the patient developed an uterovaginal prolapse due to high intra-abdominal pressure as a result of the persistent ascites, for which sacrospinal fixation was performed in December 2014. The surgery was complicated by an abdominal sepsis. The patient recovered after treatment with antibiotics.
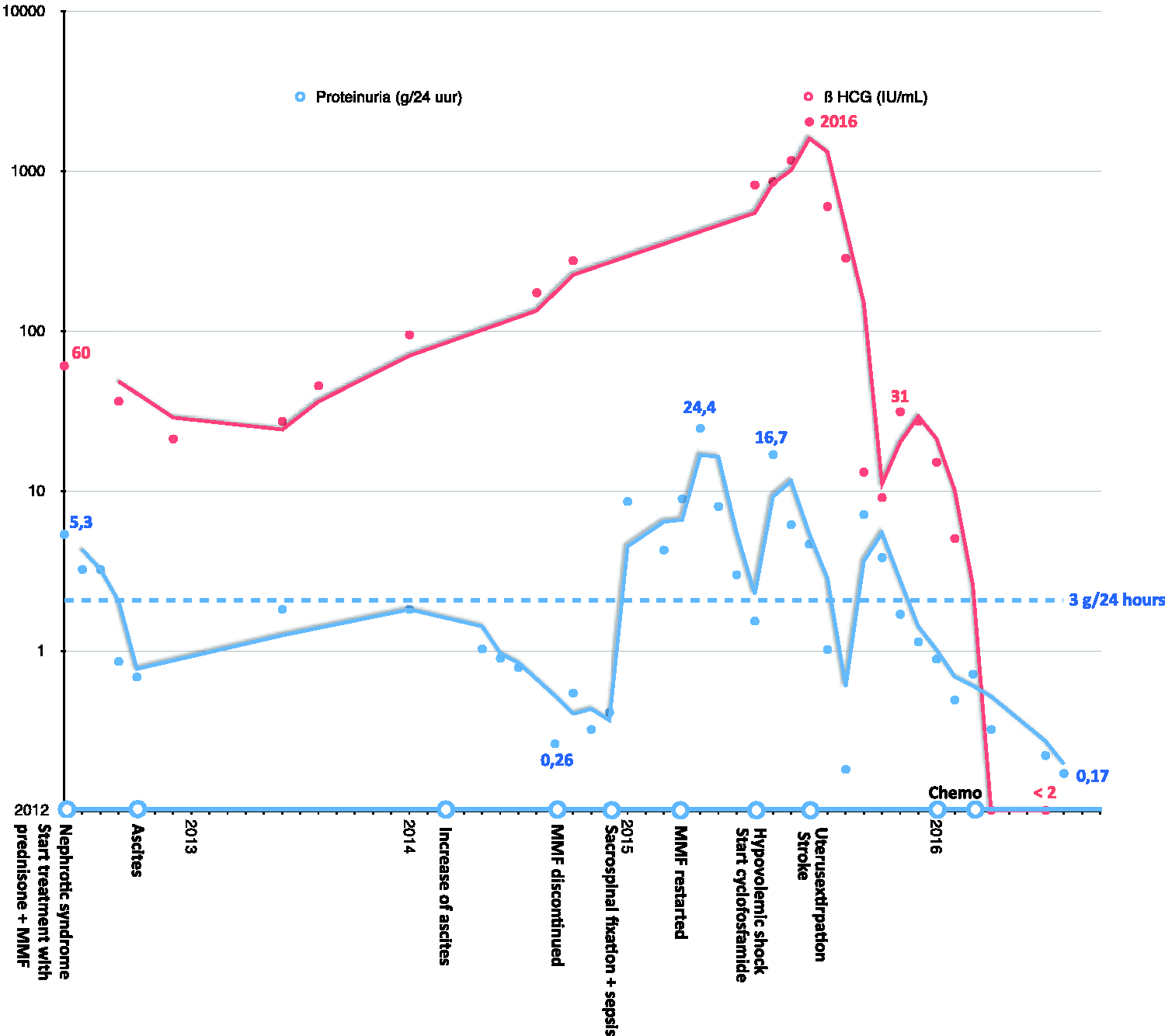
Time course showing β-HCG levels and proteinuria with symptoms and treatments on the x-axis.
In October 2014 MMF was discontinued due to drug-induced biopsy-proven liver damage. Several months later the nephrotic syndrome relapsed (proteinuria 8.51 g/24h) after which a second kidney biopsy was performed. This revealed similar findings compared to the biopsy of 2012, suggestive for either lupus nephritis or cryoglobulinemic glomerulonephritis (Figure 1). Blood tests were negative for cryoglobulins, however ANA were present and also antibodies against Sm and RNP antigens, but not to dsDNA. Anti-cardiolipin IgG/IgM, anti-beta-2-glycoproteine IgG/IgM and lupus anticoagulant were negative. These serological findings in combination with the histological findings of the renal biopsies were supportive for the diagnosis of systemic lupus erythematosus. The question arose whether the PLE and NRH could also be part of the same disease entity, as described previously. 11 MMF was reinstated without effect. Next, induction with cyclophosphamide (6 fortnightly pulses of 500 mg) and prednisone was administered in accordance with the Eurolupus regimen, 12 followed by azathioprine.
Over the course of 2015 the patient had recurrent transient ischemic attacks and one minor stroke. Libman-Sacks endocarditis and antiphospholipid syndrome were considered unlikely based on a normal transthoracic cardiac ultrasound and the absence of antiphospholipid antibodies.
In July 2015, the patient was admitted to the intensive care unit with signs of shock due to intra-pelvic hemorrhage of unknown source. As there were no apparent signs of malignancy on MRI, an elective hysterectomy was performed. Pathological analysis revealed a partially removed 6.4 cm PSTT. Adjuvant chemotherapy was administered according to the etoposide and cisplatin with etoposide, methotrexate and actinomycin D (EP-EMA) schedule. 13 After surgery, and subsequent chemotherapy, the nephrotic syndrome went into remission and remained so even after tapering prednisone and discontinuing azathioprine. The edema, ascites and Raynaud’s phenomenon disappeared and there were no further cerebrovascular events. Serological testing was repeated and ANA were absent. In retrospect, the PSTT appears to have been the cause of the nephrotic syndrome and lupus-like disease in this patient.
Discussion
Trophoblastic tumors are rare and develop secondary to a completed pregnancy or a hydatidiform mole. The discovery of such a tumor has been reported between several days up to 23 years after pregnancy or mola. In this patient’s case the tumor was not seen on prior repetitive imaging, but was discovered after a laparoscopy years later. Interestingly, renal biopsy showed a pattern partially compatible with lupus nephritis. Two separate renal biopsies showed capillary loops filled with PAS-positive but IgG negative depositions. This has been described in similar cases. 7 In our case, upon electron microscopy subendothelial granular electron dense deposits were found with only minimal mesangial depositions. In these renal biopsies, IgM is positive in immunofluorescent analysis which is probably due to ‘trapping’ of IgM in these pseudothrombi.
Trophoblastic tumors complicated by a nephrotic syndrome due to an illness resembling paraneoplastic systemic lupus erythematosus have been described before (Table 1). These complications are exceedingly rare. In all 8 cases hysterectomy was performed within months after onset of symptoms, leading to immediate remission of the nephrotic syndrome. In our case, although elevated β-HCG levels were found upon first presentation with nephrotic syndrome, PSTT was not discovered until years later. During this delay the patient developed multiple lupus-like symptoms such as Raynaud’s phenomenon, PLE, and NRH. 11 A 'full' lupus-like syndrome such presented in this report, has to our knowledge never been described before in relation to a PSTT.
Paraneoplastic systemic lupus erythematosus has been described previously.14–19 Expanding evidence indicates that several idiopathic autoimmune diseases can be early manifestations of occult cancer. 17 Though the etiology is unclear, it has been hypothesized that the break of self-tolerance against (tumor) antigens may lead to an antigen-driven immune activation with the formation of auto-antibodies leading to a syndrome that is indistinguishable from “true” systemic lupus erythematosus. 14 Another hypothesis proposes that toxins produced by tumor cells trigger the autoimmune reaction. 17 Generally preceding the diagnosis of malignancy, these auto immune diseases could be predictive of cancer and aid early diagnosis and treatment when recognized as paraneoplastic manifestations.
In our case, a diagnostic delay resulted from a phenomenon called anchoring bias 20 : all physicians assumed the patient had SLE without questioning this diagnosis in the light of the unusual and unexplained finding of increased tumor markers. Connecting these two rare clinical entities might have led to an earlier correct diagnosis. Apparently the patient herself had felt for years that her symptoms were related to the increased tumor markers. When we informed her about our conclusions and asked for permission to write this report, she explained that she had asked for a hysterectomy on multiple occasions. This request was however not honored by her physicians because imaging was repeatedly negative. Eventually, a detrimental clinical situation resulted in a laparoscopy which finally reversed the anchoring bias and led to the correct diagnosis. The patient explained to us that eventhough the resection had been irradical, she had decided not to have a re-resection because she was tired and done with hospitals after years of treatment and hospitalizations. She hoped this report would remind other physicians to keep an open mind concerning their patient’s assumed diagnosis at all times in order to avoid premature diagnostic closure. Assumed chronic diseases are notorious for this diagnostic pitfall 21 and due to the complexity it happens regularly in the case of presumed SLE. 22
In conclusion, PSTT rarely causes an SLE-like syndrome, but our case illustrates that physicians should keep an open mind concerning their patient’s assumed diagnosis at all times in order to avoid premature diagnostic closure.
Footnotes
Final diagnosis
Lupus-like paraneoplastic syndrome in a placental site trophoblastic tumor.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent to publish this information was obtained from the patient.
Contributorship
SL, JB and MH reviewed the patients history as well as the existing literature and were major contributors in writing the manuscript. JK and JR performed the histological examination of the kidney. LJ was directly involved as the patients main physician in patient care and contributed to drafting the manuscript. MO revised the manuscript. All authors read and approved the final manuscript.
Availability of data and materials
Not applicable.
Acknowledgements
Not applicable.