
Abstract
Objective
To evaluate the safety, pharmacokinetics, pharmacodynamics, and exploratory efficacy of amiselimod, an oral selective sphingosine 1-phosphate receptor-1 modulator, in patients with systemic lupus erythematosus (SLE).
Methods
A multicenter, open-label phase Ib trial was conducted in Japan. Patients in Part 1 and Part 2-B received 0.2 mg amiselimod while those in Part 2-A received 0.4 mg amiselimod for 24 weeks.
Results
Seventeen subjects received 0.2 or 0.4 mg amiselimod. Amiselimod and amiselimod-P plasma concentrations increased dose-dependently. Peripheral blood lymphocyte count decreased in all patients after amiselimod treatment, with no clear dose response. There were no serious/severe adverse events (AEs) or clinically meaningful cardiac effects. Five subjects were withdrawn from amiselimod treatment following a decrease in lymphocyte count to <200/μl. Anti-double stranded-DNA antibody decreased from baseline to Week 24/end of treatment (EOT), with those in 2 subjects (22.2%) decreasing to within the normal range. Total SLE disease activity index 2000 score decreased by ≥4 at EOT in 7 of 17 subjects.
Conclusions
Amiselimod was generally well tolerated. While no serious AEs or infectious AEs led to discontinuation, low lymphocyte counts of <200/μl were observed as a laboratory abnormality. Our findings suggest the potential efficacy of amiselimod for patients with SLE.
Trial registration: ClinicalTrials.gov identifier: NCT02307643.
Keywords
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by the development of heterogeneous autoantibodies and subsequent tissue damage. The pathogenesis of SLE is thought to involve the production of autoantibodies following the activation of autoreactive T and B cells, deposition of immune complexes in target tissues and organs, and invasion by autoreactive T cells. Currently, belimumab, which inhibits B lymphocyte stimulation, is the only approved biological agent for SLE. However, the efficacy of drugs targeting B cells is limited, and other approaches, including targeting of T cells, are required to improve treatment options for SLE patients.1–3
Sphingosine 1-phosphate (S1P) and the S1P1 receptor are essential for lymphocyte egress from secondary lymphoid organs. S1P1 receptor modulators decrease peripheral lymphocyte counts by inhibiting lymphocyte egress from secondary lymphoid organs, and subsequently inhibit infiltration of autoreactive T cells into sites of inflammation.4,5 The S1P1 receptor modulators fingolimod, 6 siponimod 7 and ozanimod 8 have demonstrated clinical efficacy for multiple sclerosis, and these agents are expected to be applicable to other autoimmune diseases.
Studies in SLE animal models indicate that S1P1 receptor modulators inhibit lupus nephritis progression, increase the survival rate,9–13 and decrease infiltration of T cells and macrophages into the kidneys. 12 Two compounds (KRP-203 and cenerimod) have been trialed for the treatment of patients with lupus, with a 12-week treatment with cenerimod showing promising therapeutic efficacy.14,15 These findings suggest the therapeutic potential of S1P1 modulators for SLE.
Amiselimod (MT-1303), an oral selective S1P1 receptor modulator, is converted in vivo to its active metabolite, amiselimod phosphate (amiselimod-P), 16 and has a more favorable cardiac safety profile than fingolimod. 17 Our previous findings confirmed that amiselimod inhibits lupus nephritis progression in MRL/lpr and NZBWF1 mice, 18 suggesting its potential effectiveness in the treatment of SLE in humans.
SLE patients generally exhibit lower lymphocyte counts than healthy subjects; however, safety information regarding administration of S1P1 receptor modulators with respect to the reduced lymphocyte count in SLE patients is insufficient. Additionally, although combination therapies comprising corticosteroids and immunosuppressants are common in the treatment of SLE, safety information regarding combined treatment with amiselimod in patients with SLE and other diseases is lacking. Here, we conducted an open-label exploratory study to evaluate the safety, pharmacokinetics, pharmacodynamics, and exploratory efficacy of amiselimod in SLE patients, including tolerability when concomitantly administered with corticosteroids and immunosuppressants.
Materials and methods
Study design
The study was conducted under an open-label, non-randomized multicenter phase 1b design. The primary objective was to evaluate the safety of amiselimod in SLE patients, and the secondary objectives were to assess the drug’s pharmacokinetics (PK) and pharmacodynamics (PD) and its effect on several biological markers, and to explore its exploratory clinical efficacy. The study was conducted at 9 medical institutions in Japan between February 2015 and May 2017 in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice guidelines (NCT02307643). All subjects provided written informed consent to participate. Prior to the conduct of the study, the protocol was reviewed and approved by the institutional review board of each of the participating institutions.
After a screening period, subjects were entered into a 24-week treatment period, followed by a 12-week safety follow-up period. The study comprised 3 parts: in Part 1, patients on low doses of corticosteroids received amiselimod at an oral dose of 0.2 mg; in Part 2-A, patients on low doses of corticosteroids received amiselimod at an oral dose of 0.4 mg; and in Part 2-B, patients on low doses of corticosteroids and immunosuppressant agents received amiselimod at an oral dose of 0.2 mg. Part 2-A and 2-B were started after first confirming changes in blood lymphocyte counts and adverse events (AEs) 4weeks after the start of treatment in the final subject of Part 1. The planned number of subjects for treatment with amiselimod was 18, with 6 subjects in each part.
Study population
Patients were aged 20 to <65 years and had been diagnosed with SLE using the criteria of the American College of Rheumatology (ACR) (1997) or revised criteria of the Systemic Lupus International Collaborating Clinics (SLICC) (2012).19,20 Disease activity in SLE was determined by the presence of at least one of the following items at 2 time points during the screening period: 1) positive for anti-double stranded (ds)-DNA antibodies; 2) low complement levels (complement component 3 [C3], complement component 4 [C4], or 50% hemolytic complement [CH50] levels below the lower limit of normal); and 3) systemic lupus erythematosus disease activity index 2000 (SLEDAI-2K) score ≥4. Patients who were treated with stable doses of corticosteroids (equivalent to ≤15 mg/day of prednisolone) for at least 4 weeks before screening were entered into the study. Dose tapering was permitted at the discretion of the investigator from 12 weeks after the start of amiselimod treatment through to the end of the follow-up period. In Part 1 and Part 2-A, antimalarial drugs, immunosuppressants, intravenous immunoglobulin and biologic agents were prohibited. In Part 2-B, only one of the following medications was required at a steady dose: ≤ 2 mg/kg/day of azathioprine, ≤ 3 3mg/day of tacrolimus, ≤150 mg/day of mizoribine, or ≤16 mg/week of methotrexate. Since neither anti-malarial drugs nor mycophenolate mofetil were approved for the treatment of SLE in Japan at the time of study initiation, their concomitant use in this study was not permitted. Patients with severe active lupus nephritis, active neuropsychiatric SLE or thrombosis were excluded from the study, as were patients with white blood cell counts of <3,000/μl and lymphocyte counts during the screening period of <800/μl. Patients with cardiovascular diseases or clinically significant abnormal findings in the 12-lead electrocardiogram (ECG) or Holter ECG at screening were also excluded.
PK and PD evaluation
Plasma concentrations of amiselimod and amiselimod-P were measured using a fully validated liquid chromatography-tandem mass spectrometry method (LGC Limited). Peripheral lymphocyte counts and lymphocyte subsets were determined in the central laboratory (LSI Medience Corporation, Tokyo, Japan) as PD evaluation. The following lymphocyte subsets were analyzed: T cells (CD3+), T helper type (Th) 1 cells (CD4+/CD45RA-/CXCR3+/CCR4-/CCR6-), Th17 cells (CD4+/CD45RA-/CXCR3-/CCR4+/CCR6+), B cells (CD19+), plasmablast cells (IgD-/CD27++/CD38++/CD138-), and plasma cells (IgD-/CD27++/CD38++/CD138+).
In addition, as a safety evaluation, peripheral lymphocytes were counted in the local laboratory to determine whether to continue or discontinue treatment with amiselimod.
Safety evaluation
Biochemistry and urine assessments were conducted at the central laboratory. Hematology assessment was conducted in the local laboratory as a quick safety assessment at each visit. For all other safety assessments, vital signs assessment, 12-lead ECG, Holter ECG, and optical coherence tomography were performed.
Measurement of biomarkers
Anti-ds-DNA IgG antibody titer and serum complement (C3, C4, CH50) levels were measured at the central laboratory as biomarkers in SLE.
Clinical assessment
At screening, Week 4, Week 12, Week 24 and follow-up (FU)-Week 12, the clinical outcome measures for disease activity, SLEDAI-2K, 21 British Isles Lupus Assessment Group (BILAG)-2004 index,22,23 and physician’s global assessment (PGA), were evaluated. For SLEDAI-2K, a reduction of ≥4 from the score at baseline was used to indicate a clinically meaningful improvement according to previous studies.24,25 Patients with BILAG-2004 activity were defined as those with BILAG grade A activity in ≥1 or BILAG grade B activity in ≥2 organ systems, in accordance with recent clinical studies. 25 For PGA, an increase of <0.3 was considered to indicate the absence of worsening disease activity.
Statistical analyses
We examined 6 subjects in each part because this is the number of subjects required for safety evaluation in an exploratory study at the early stages of clinical development. In safety, efficacy and PD analyses, all subjects were analyzed according to the part of the study to which they were registered. The baseline value was the last observed value of the parameter of interest prior to the first intake of amiselimod. No observation was inputted in the event of missing data. Values obtained at the end of treatment (EOT) visit were defined as the observed values at Week 24 in subjects who completed the planned treatment or the last observed values in subjects who prematurely discontinued the planned treatment. Statistical analysis and interval estimation were not conducted. All adverse events were coded according to the terms used in the Medical Dictionary for Regulatory Activities (MedDRA).
Results
Subject disposition and demographics and other baseline characteristics
A total of 17 subjects received amiselimod 0.2 or 0.4 mg. Six subjects (35.3%) were withdrawn from the study during the treatment period, while 66.7% of subjects in Part 1 and Part 2-B and 60.0% of subjects in Part 2-A completed the planned treatment (Figure 1). Among the 6 withdrawn subjects, 5 met the discontinuation criteria for low lymphocyte count <200/μl, consisting of 2 subjects from Part 1, 1 from Part 2-A and 2 from Part 2-B, while the 6th was withdrawn because the investigator judged it was necessary to follow up the subject’s hepatic function test.
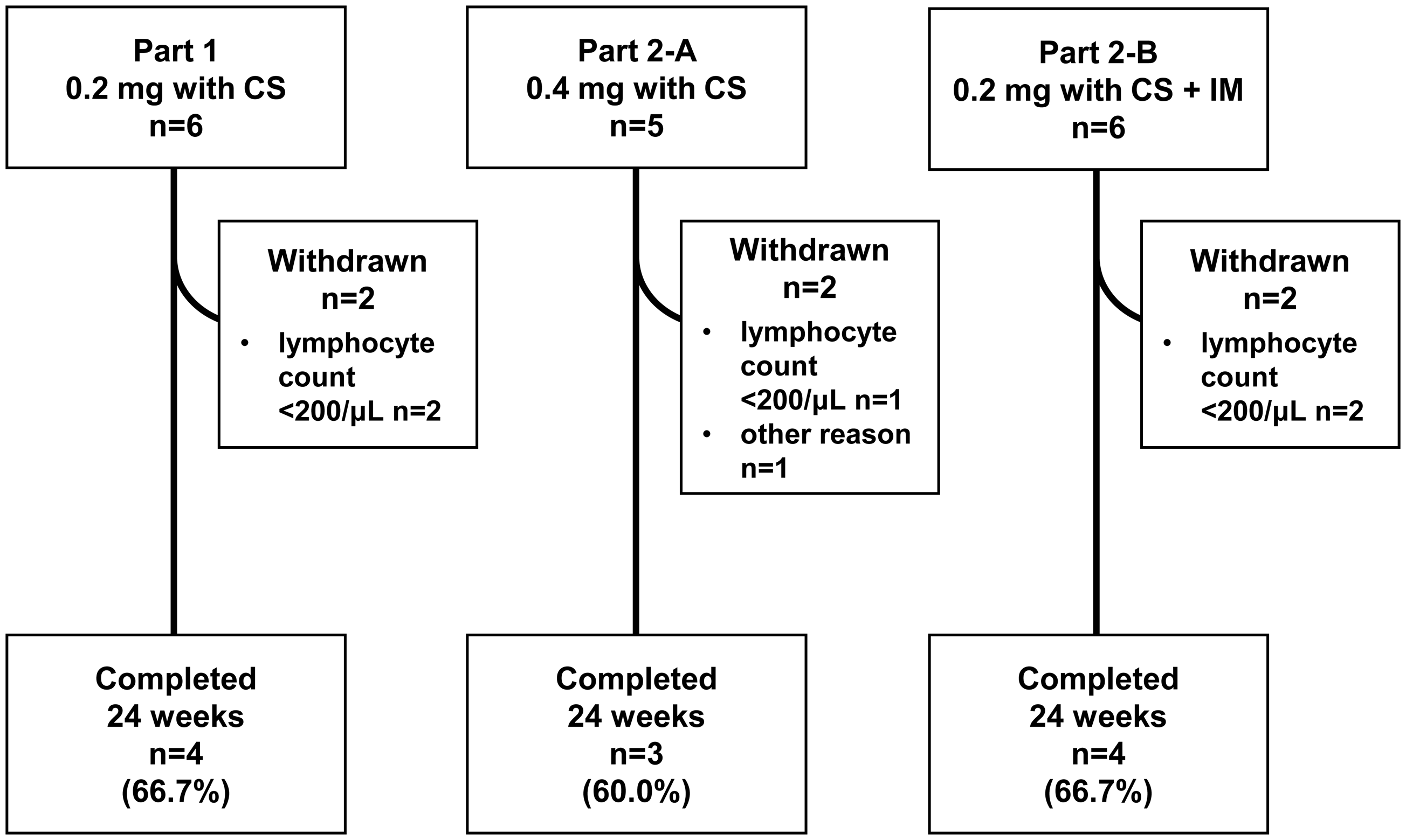
Patient disposition.
Demographic and other baseline characteristics are summarized in Table 1. The baseline anti-ds-DNA antibody level, SLEDAI-2K score and corticosteroid dosage in Part 1 were slightly lower than the respective values in Part 2-A and Part 2-B.
Demographics and other baseline characteristics.
Note: The “overall” column includes data from subjects from all three study parts. In Part 2-B, all subjects used immunosuppressants as concomitant medication. The most frequently used immunosuppressant was tacrolimus (3/6, 50.0%), followed by azathioprine (2/6, 33.3%) and mizoribine (1/6, 16.7%).
Anti-ds-DNA antibody: anti-double stranded deoxyribonucleic acid antibody; C3: complement 3; C4: complement 4; CH50: 50% hemolytic complement activity; SLE: systemic lupus erythematosus; SLEDAI-2K: SLE Disease Activity Index 2000.
PK and PD (lymphocyte count) evaluation
The concentration of amiselimod and amiselimod-P in plasma increased in a dose-dependent manner (Supplementary Figure 1). Mean half-life of amiselimod and amiselimod-P ranged from 499 to 564 hours and 465 to 534 hours, respectively. A decrease in peripheral lymphocyte count was observed in all subjects after treatment with amiselimod. A marked decrease in lymphocyte count was observed by Week 4 and remained at a nadir thereafter during the treatment period (up to Week 24). The mean (minimum, maximum) lymphocyte count at Week 4 was 0.413 (0.27, 0.64) × 109/l in Part 1, 0.368 (0.14, 0.55) × 109/l in Part 2-A, and 0.438 (0.27, 0.60) × 109/l in Part 2-B, which corresponds to 39% (21%, 55%), 40% (8%, 72%), and 47% (30%, 63%), respectively. The mean lymphocyte count for all 3 study parts returned to 80% or more of baseline after the cessation of amiselimod administration (Figure 2(a)). The mean lymphocyte count at Week 24 was 0.320, 0.430 and 0.385 × 109/l for subjects in Part 1, Part 2-A and Part 2-B, which corresponds to 30%, 46% and 42% of baseline, respectively. Amiselimod 0.2 mg (Part 1 and Part 2-B) and 0.4 mg (Part 2-A) showed numerically similar PD effects. The number of subjects with a lymphocyte count <0.2 × 109/l (measured in the central laboratory) during the 24-week treatment period was 1 (16.7%), 2 (40.0%) and 1 (16.7%) among subjects in Part 1, Part 2-A and Part 2-B, respectively.
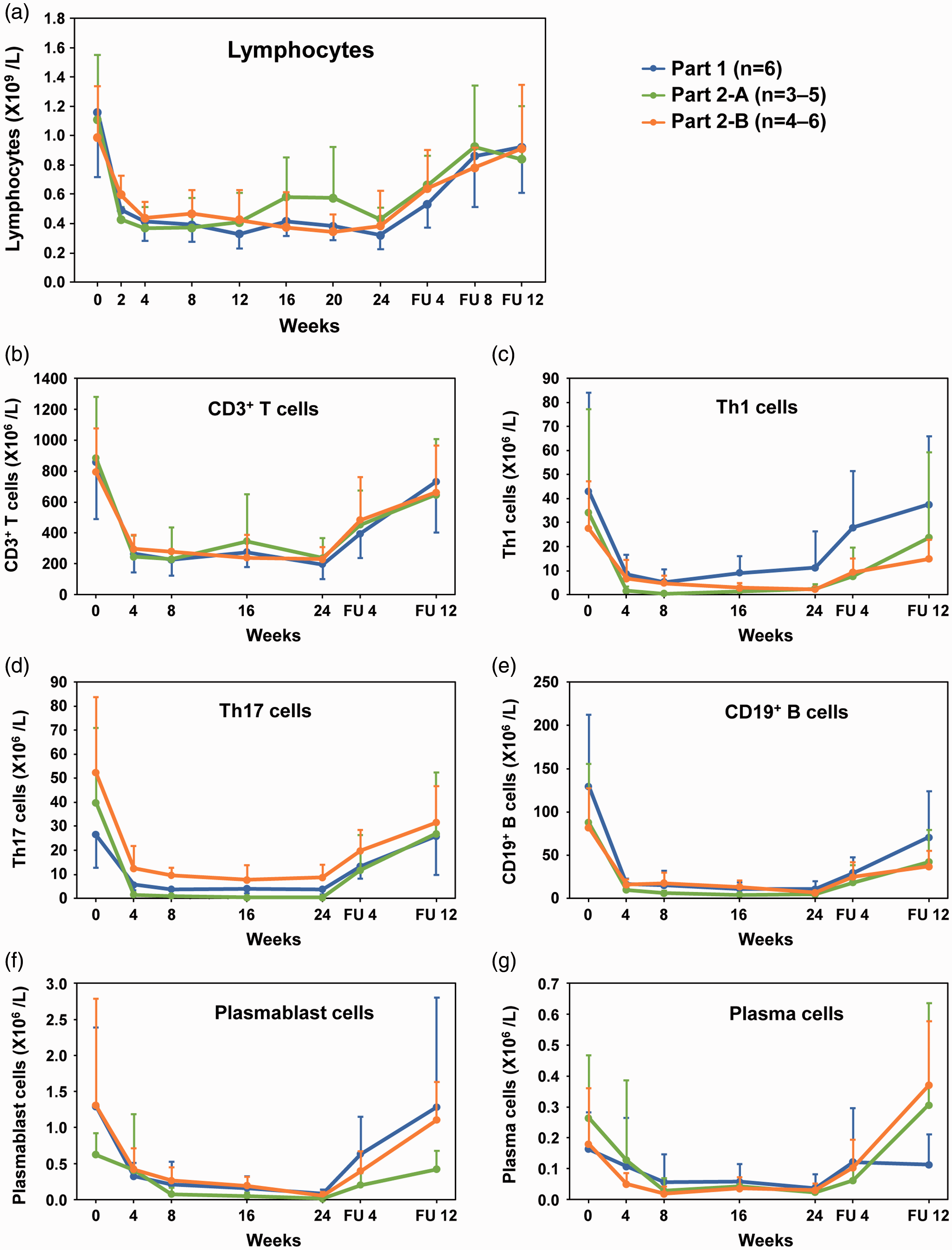
Number of peripheral lymphocytes.
Figure 2(b) to (g) show the mean counts of T, Th1, Th17, B, plasmablast and plasma cells in peripheral blood at each time point. The decreasing trend in the change in the number of respective lymphocyte subsets was similar to that of the total lymphocyte count. No clear dose-response was observed in this study.
Safety
The mean (SD) number of days of exposure to amiselimod was 169.0 (5.83), 122.0 (62.57) and 138.2 (57.87) among subjects in Part 1, 2-A and 2-B, respectively.
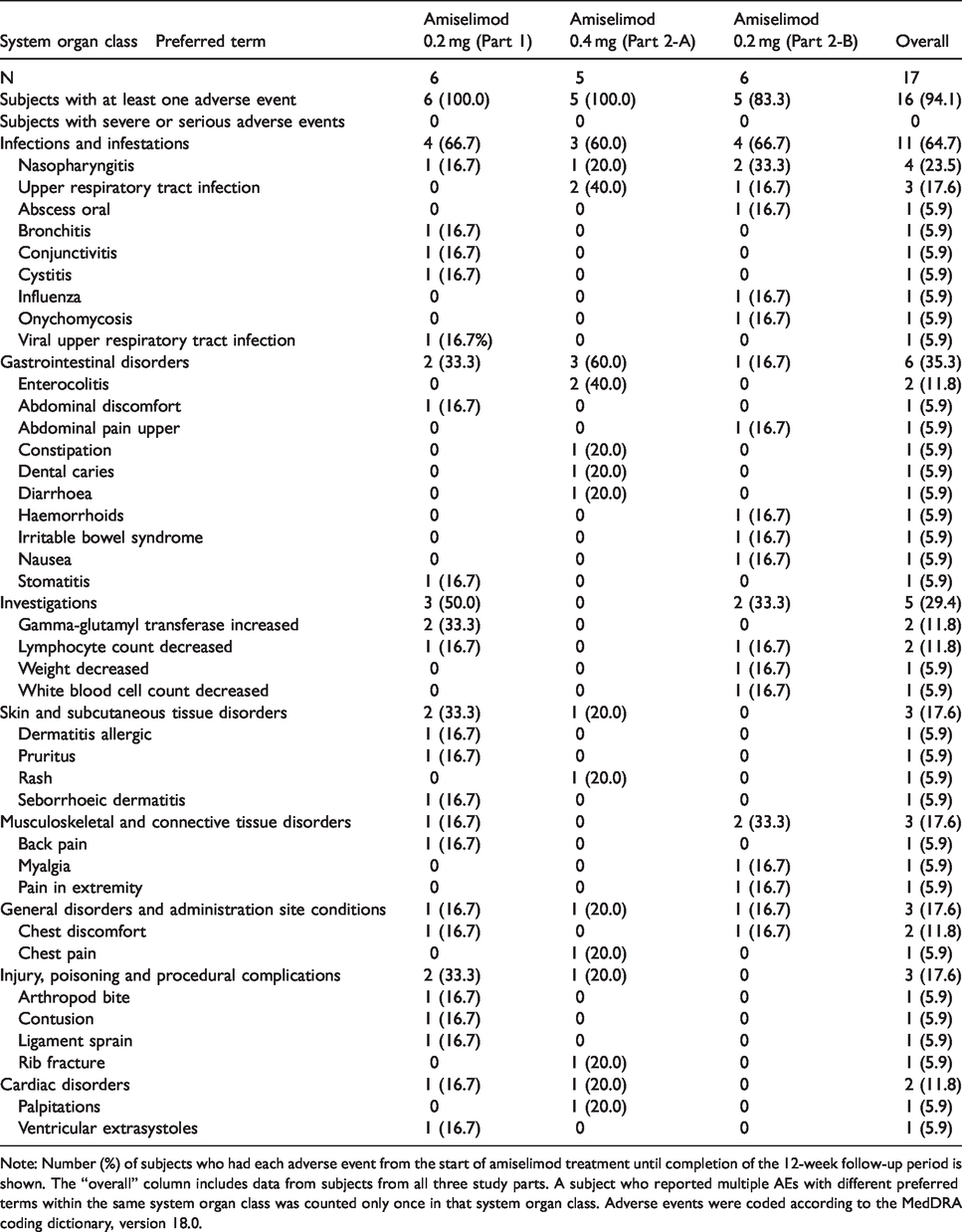
There were no deaths, serious AEs, severe AEs or AEs related to infection that led to the discontinuation of amiselimod in this study. One or more AEs were reported by 6 (100.0%), 5 (100.0%), and 5 (83.3%) subjects in Part 1, 2-A and 2-B, respectively (Table 2). The most commonly reported preferred term for AEs was nasopharyngitis (4/17, 23.5%), followed by upper respiratory tract infection (3/17, 17.6%). Most reported AEs were mild. No obvious differences in AEs were observed among the study parts, and there was no trend in AE incidence across increasing amiselimod doses or in combination with immunosuppressants.
Adverse events in >1 subject according to system organ class.
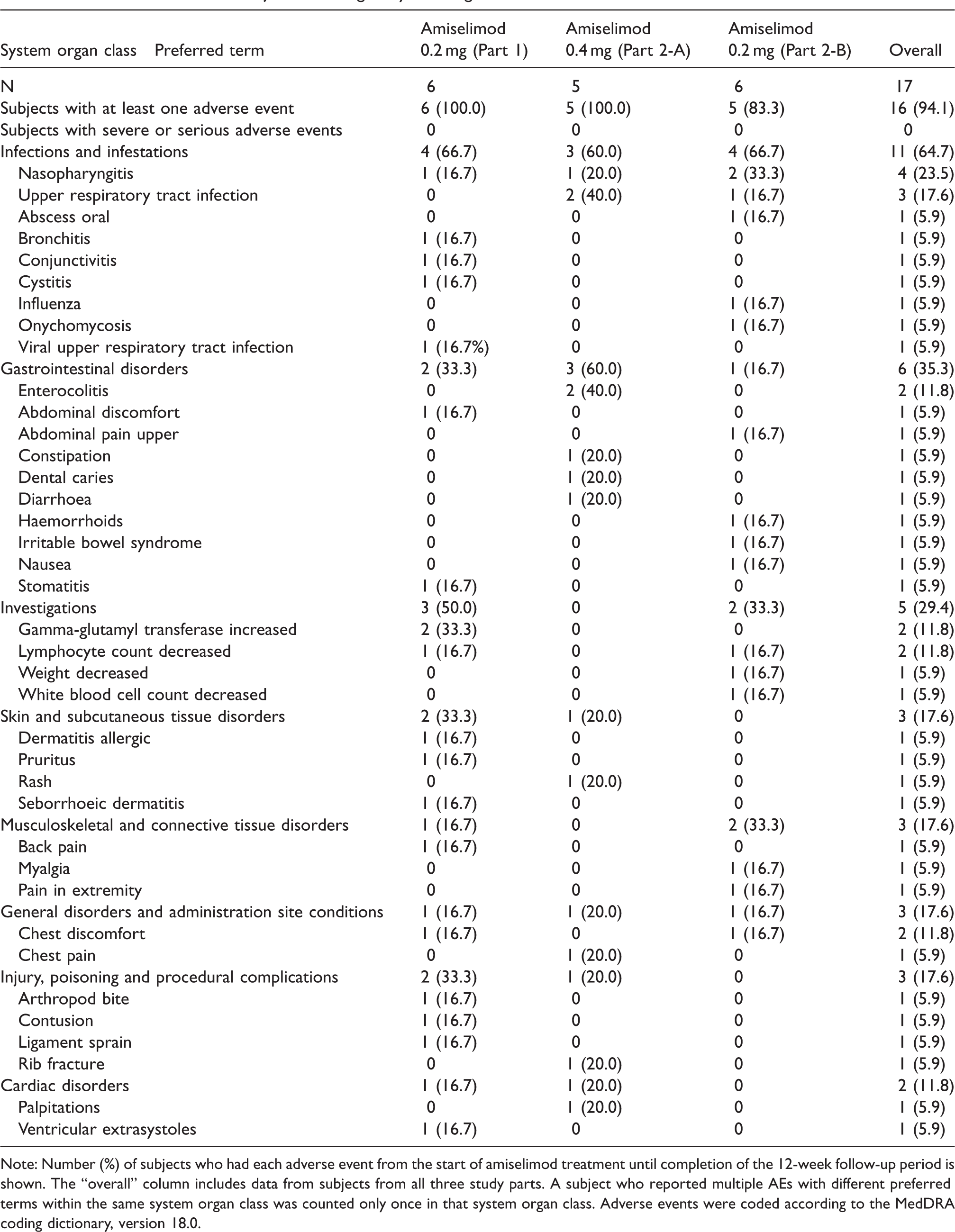
Note: Number (%) of subjects who had each adverse event from the start of amiselimod treatment until completion of the 12-week follow-up period is shown. The “overall” column includes data from subjects from all three study parts. A subject who reported multiple AEs with different preferred terms within the same system organ class was counted only once in that system organ class. Adverse events were coded according to the MedDRA coding dictionary, version 18.0.
Notable safety factors associated with S1P1 receptor modulators include decreased lymphocyte count and decreased heart rate. 6 S1P1 receptor modulators decrease peripheral lymphocyte counts, and patients with SLE are known to have lower lymphocyte counts due to the disease itself or treatment with immunosuppressive agents. In this study, although 5 subjects met the criteria for discontinuation (low lymphocyte count of <200/μl) and were withdrawn from treatment, no serious AEs were observed in these 5 subjects. Infectious AEs (severity) that occurred within 1 month before and after measurement of a low lymphocyte count of <200/μl were cystitis (mild), nasopharyngitis (mild) and oral abscess (moderate). To assess whether a reduction in lymphocyte count was considered an AE, the physician evaluated whether the decrease in lymphocyte count was significant based on each subject’s background. Low lymphocyte count of <200/μl was reported as an AE in 2 of 5 subjects.
No clinically significant bradycardia events were observed during the study, and no clinically significant abnormalities were observed in the 12-lead ECG except for mild ventricular extrasystoles at Week 20 in one subject in Part 1.
Regarding the clinical laboratory evaluations, 2 subjects in Part 1 experienced elevated γ-glutamyl transpeptidase, and 1 subject in Part 2-A experienced abnormal hepatic function accompanied by elevated ALT, AST and γ-glutamyl transpeptidase. No subjects met the criteria for Hy’s law (ALT ≥3 × upper limit of normal [ULN] or AST ≥3 × ULN, and total bilirubin ≥2 × ULN). There were no other safety findings of clinical concern, as measured by vital signs and OCT assessments.
Changes in biomarkers (anti-ds-DNA antibody and complements)
Nine subjects (52.9%) across all study parts had positive levels (>12 IU/ml) of anti-ds-DNA antibody at baseline (Table 1). Of the 9 subjects, anti-ds-DNA antibody levels in 8 (88.9%) decreased from baseline to Week 24/EOT (Figure 3(a)) and those in 2 (22.2%) decreased to less than 12 IU/ml (within the normal range) at Week 24/EOT (27.3 to 10.3 IU/ml, 25.7 to 7.1 IU/ml).
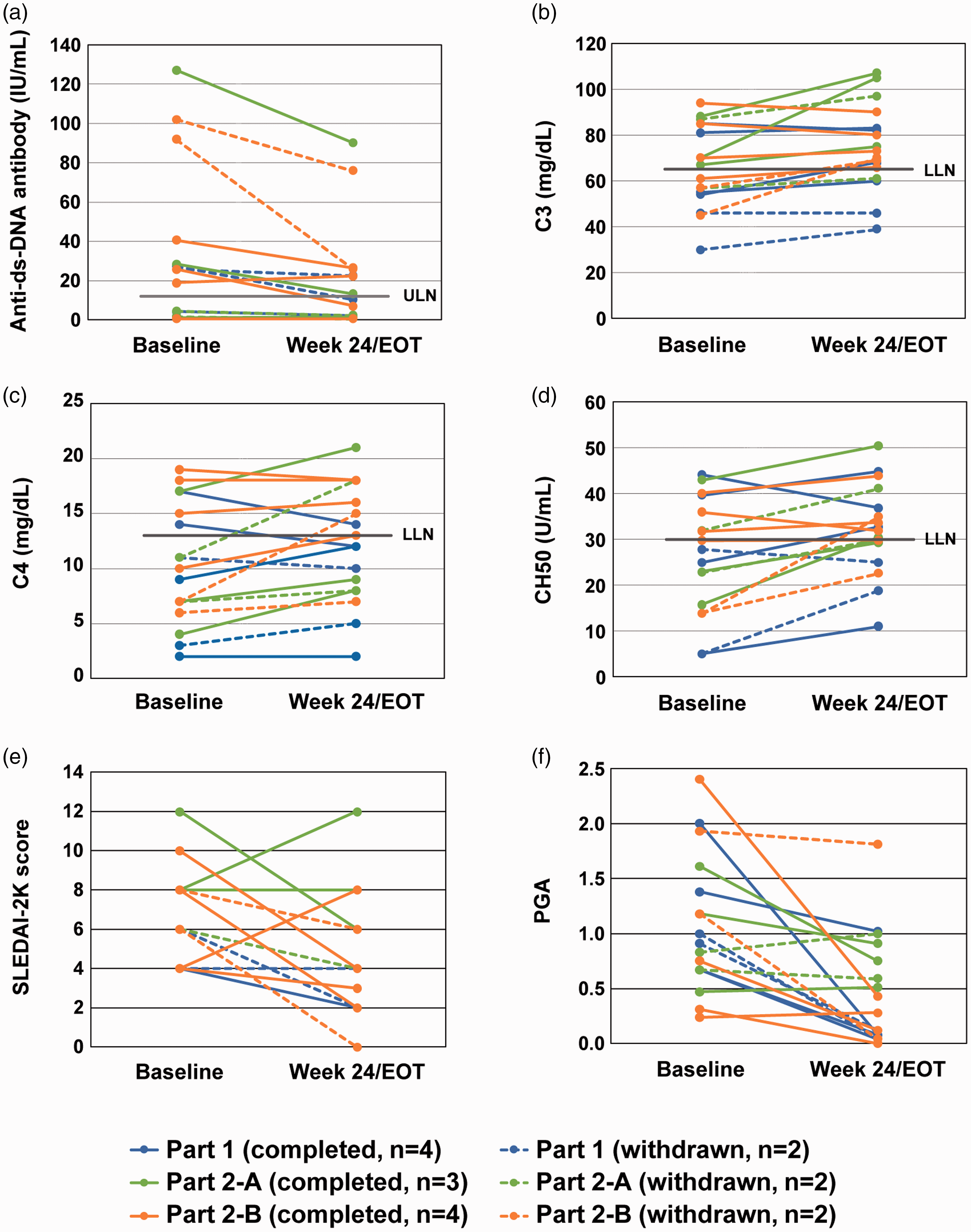
Individual biomarkers (anti-ds-DNA antibody and complements) and clinical assessment (SLEDAI-2K and PGA).
Low levels of C3, C4, and CH50 were observed in 8/17 (47.1%), 11/17 (64.7%), and 10/17 (58.8%) subjects, respectively (Table 1). Four of 8 (50.0%), 3 of 11 (27.3%) and 4 of 10 (40.0%) subjects with levels of C3, C4 and CH50 lower than the normal range at baseline showed an increase in levels to above the lower limit of normal at Week 24/EOT compared to that at baseline. The number (percentage) of subjects with increased (>0) complement levels from baseline among those with lower levels than the normal range at baseline was 7/8 (87.5%) for C3, 9/11 (81.8%) for C4 and 9/10 (90.0%) for CH50 at Week 24/EOT (Figure 3(b) to (d)). There were no obvious differences among the three study parts.
Summary of clinical assessment
The median SLEDAI-2K score across all three study parts was 6.0 and the score was ≥4 for all subjects (Table 1). All except one patient met the disease activity criteria of SLEDAI-2K ≥4 at 2 time points during the screening period. The mean SLEDAI-2K score decreased from baseline to both Week 24 and EOT in each study part (Table 3). The individual SLEDAI-2K score decreased from baseline to Week 24/EOT in 13 of 17 subjects (Figure 3(e)). The SLEDAI-2K score of 7 of 17 subjects was reduced by ≥4 points from baseline to EOT. The number of subjects with BILAG grade A activity in ≥1 or BILAG grade B activity in ≥2 organ systems decreased from baseline to both Week 24 and EOT in each study part. In the PGA, no subjects showed a > 0.3 increase from baseline to either Week 24 or EOT in any study part (Table 3, Figure 3(f)).
Summary of clinical assessment.
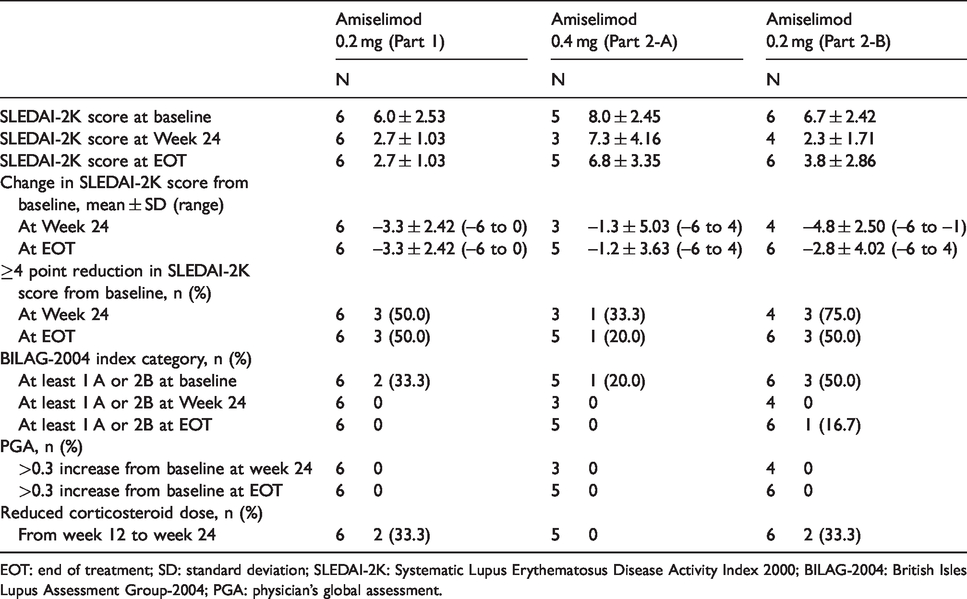
EOT: end of treatment; SD: standard deviation; SLEDAI-2K: Systematic Lupus Erythematosus Disease Activity Index 2000; BILAG-2004: British Isles Lupus Assessment Group-2004; PGA: physician’s global assessment.
Regarding SLE-related symptoms, improvements were observed in arthritis, alopecia, rash, and mucosal ulcer, which are listed as SLEDAI-2K symptoms. The number of subjects with each symptom at baseline and EOT was 7/17 (41.2%) and 2/17 (11.8%) for arthritis, 8/17 (47.1%) and 6/17 (35.3%) for alopecia, 9/17 (52.9%) and 3/17 (17.6%) for rash, and 3/17 (17.6%) and 0/17 (0%) for mucosal ulcer, respectively.
Corticosteroid tapering was allowed in this study from 12 weeks after the start of treatment. Four of 17 subjects reduced their corticosteroid dosage between Week 12 and Week 24 during the treatment period. One of these 4 subjects increased their corticosteroid dosage during follow-up because of disease flare.
Discussion
Our primary objective in this phase 1b study was to evaluate the safety of amiselimod in patients with SLE. Oral treatment with amiselimod for 24 weeks was generally well tolerated. There were no deaths, serious AEs, severe AEs, or infectious AEs that led to discontinuation of amiselimod. There were likewise no clinically meaningful cardiac effects, including bradycardia, and the safety profile of amiselimod in this study was consistent with that in previous reports.16,17,26 Our findings therefore indicate that amiselimod is tolerable in patients with SLE when concomitantly administered with corticosteroids and immunosuppressants.
Amiselimod and amiselimod-P had similarly long half-lives around 450 to 550 hours. This result is consistent with previous reports, and this pharmacokinetic profile is considered beneficial for little cardiac effects without a titration regimen. 17
Five subjects (30%) treated with amiselimod 0.4mg or 0.2 mg were withdrawn prematurely based on the discontinuation criterion (confirmed lymphocyte count of <200/μl), which was set based on the guidance for clinical trials for fingolimod in MS. 6 The mean lymphocyte count was 320–430/μl among subjects in each study part during the treatment period with amiselimod and was lower than that in MS patients. However, the mean percent reduction in lymphocyte count from baseline (approximately 54–70% reduction) in each study part did not markedly differ from that for MS. Although a previous phase 3 study on fingolimod reported that there was no clear association between a low lymphocyte count of <200/μl and an increase in infection rate, 27 a prolonged decrease in lymphocyte count is likely a risk for infection and infection is a leading cause of morbidity and mortality in SLE. Accordingly, further investigation is required to determine the relationship between a prolonged decrease in lymphocyte count and infection in patients taking amiselimod. With regard to the relationship between dose and lymphocyte count reduction, a dose-dependent reduction in lymphocyte count following administration of amiselimod, as observed in MS patients, 26 was not observed in the present study. We predict that the lack of an observed dose dependency may be attributed to the limited number of registered patients, which was further reduced by the withdrawal of patients with a lymphocyte count of <200/μl. Alternatively, it is possible that 0.2mg amiselimod may be sufficient to produce sub-maximal effects in SLE patients, suggesting that doses below 0.2 mg (e.g., 0.1 mg) may significantly reduce lymphocyte counts in SLE patients.
Efficacy assessment using biomarkers and clinical assessment suggest that amiselimod treatment may be effective for SLE patients. We observed a decrease in the proportion of subjects with skin and joint symptoms according to SLEDAI-2K, with more than 50% of subjects who completed the 24-week treatment showing a ≥ 4-point reduction in their SLEDAI-2K score. We also observed a decrease in the proportion of subjects with BILAG grade A activity in ≥1 or BILAG grade B activity in ≥2 organ systems. In addition, there was a decrease in the number of Th1 and Th 17 cells in peripheral blood following administration of amiselimod. Patients with SLE or lupus nephritis have increased levels of T cells that produce inflammatory cytokines, such as interleukin-17 and interferons, in target tissues, which play an important role in the pathology of SLE.3,28–30 The reduction in these T cells in peripheral blood by amiselimod may underlie the improvement in clinical manifestations in SLE patients.
In the mouse antibody response, fingolimod reportedly reduces the production of high affinity, class-switched antibodies by reducing formation of the germinal center in the T cell-dependent antibody formation system. 31 Additionally, treatment with fingolimod reduces the specific antibody production response to immunization with anticipated novel antigens or recall antigens in MS patients. 32 The effects of amiselimod on blood anti-ds-DNA antibody levels in SLE animal models of SLE were contradictory. 18 Administration of amiselimod suppressed the increase in anti-dsDNA antibody levels in MRL/lpr but not NZBWF1 mice. However, these results are consistent with those observed with other S1P1 receptor modulators in MRL/lpr and NZBWF1 mice.9–11,13 The present study showed that amiselimod reduced anti-ds-DNA antibody levels in SLE patients presumably by reducing interactions between antigen-specific Th and B cells and subsequently suppressing the antibody-producing ability. A recent study reported similar results based on SLEDAI-2K and anti-ds-DNA antibody levels following 12 weeks of treatment with another S1P1 receptor modulator, which supports our exploratory efficacy results. 15 In summary, amiselimod suppressed both autoantibody production by B cells and infiltration of T cells into sites of inflammation, and has the potential to improve SLE symptoms and biomarker levels.
Several limitations of this study warrant mention. First, the sample size was small. Second, we did not compare the efficacy of amiselimod to a placebo control. Third, the subjects were all of the same race, namely Japanese. Fourth, because data from subjects who were prematurely withdrawn from the study were included in the analyses, interpretation of the findings should be conducted with caution. In this study, 0.2 mg amiselimod showed a favorable trend towards improving several clinical endpoints and biomarkers. These findings suggest the importance of examining the efficacy of amiselimod in SLE patients receiving immunosuppressants in an add-on double-blind study at 0.2 mg or lower doses. The efficacy of amiselimod on SLE symptoms should also be confirmed using a larger sample size and plurality of races, and by comparison with a placebo control.
In summary, our results suggest that amiselimod was generally well tolerated, with no serious AEs and no infectious AEs that led to its discontinuation, although low lymphocyte counts of <200/μl were observed as a laboratory abnormality. No safety concerns unique to SLE were observed following concomitant administration of 0.2 and 0.4 mg amiselimod with corticosteroids and immunosuppressants. Our findings additionally revealed the potential efficacy of amiselimod for patients with SLE. Further investigation of amiselimod as a therapy for SLE is warranted.
Supplemental Material
sj-pdf-1-lup-10.1177_0961203320966385 - Supplemental material for Amiselimod, a sphingosine 1-phosphate receptor-1 modulator, for systemic lupus erythematosus: A multicenter, open-label exploratory study
Supplemental material, sj-pdf-1-lup-10.1177_0961203320966385 for Amiselimod, a sphingosine 1-phosphate receptor-1 modulator, for systemic lupus erythematosus: A multicenter, open-label exploratory study by Yoshiya Tanaka, Kazuoki Kondo, Ayako Ichibori, Yoshiari Yanai, Yutaka Susuta, Shinsuke Inoue and Tsutomu Takeuchi in Lupus
Footnotes
Acknowledgments
The authors would like to thank the study participants and investigators. The authors also acknowledge Kunio Sugahara, PhD, Mitsubishi Tanabe Pharma Corporation, for critically reviewing the manuscript from a non-clinical point of view. The authors also thank Heidi Tran, PhD, from DMC Corp. (![]() ) for editing drafts of this manuscript.
) for editing drafts of this manuscript.
Availability of study materials
The datasets generated and/or analysed in the current study are not publicly available due to ethical reasons (the authors recognise the risk that patients’ identities or private information may be revealed by public data disclosure due to the small sample size and the rarity of the disease studied) but are available from Mitsubishi Tanabe Pharma Corporation (rpp_mtpc@cc.mt-pharma.co.jp) or the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Y. Tanaka has received speaking fees and/or honoraria from AbbVie GK., Astellas Pharma Inc., Bristol-Myers Squibb K.K., Chugai Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Eisai Co., Ltd., Eli Lilly Japan K.K., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corp., Novartis Pharma K.K., Pfizer Japan Inc., Takeda Pharmaceutical Co., Ltd., Teijin Ltd. and YL Biologics Ltd.; and has received research grants from Asahi Kasei Pharma Corp., Bristol-Myers Squibb K.K., Chugai Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Eisai Co., Ltd., Mitsubishi Tanabe Pharma Corp., Ono Pharmaceutical Co., Ltd., Sanofi K.K., Takeda Pharmaceutical Co., Ltd. and UCB Japan Co., Ltd. K. Kondo, A. Ichibori, Y. Yanai, Y. Susuta and S. Inoue are employees of Mitsubishi Tanabe Pharma Corporation. T. Takeuchi has received grants from AbbVie GK., Asahi Kasei Pharma Corp., Astellas Pharma Inc., AYUMI Pharmaceutical Corp., Chugai Pharmaceutical Co, Ltd., Daiichi Sankyo Co., Ltd., Eisai Co., Ltd., Mitsubishi Tanabe Pharma Corp., Nippon Kayaku Co., Ltd., Novartis Pharma K.K., Pfizer Japan Inc., and Takeda Pharmaceutical Co., Ltd.; speaking fees from AbbVie GK., Astellas Pharma Inc., Bristol-Myers Squibb K.K., Chugai Pharmaceutical Co, Ltd., Daiichi Sankyo Co., Ltd., Eisai Co., Ltd., Mitsubishi Tanabe Pharma Corp, Novartis Pharma K.K., Pfizer Japan Inc., Sanofi K.K., Takeda Pharmaceutical Co., Ltd., and Teijin Pharma Ltd.; and consultant fees from AbbVie GK., Astellas Pharma Inc., Astra Zeneca K.K., Chugai Pharmaceutical Co, Ltd., Eli Lilly Japan K.K., GlaxoSmithKline K.K., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corp, Nippon Kayaku Co., Ltd., Novartis Pharma K.K., Taiho Pharmaceutical Co., Ltd., Taisho Toyama Pharmaceutical Co., Ltd., and UCB Japan Co., Ltd.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by Mitsubishi Tanabe Pharma Corporation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.