
Abstract
Introduction
C1q is an essential part of the classical pathway of complement activation. Genetic deficiencies, caused by homozygous mutations in one of the C1q genes, are rare and are strongly associated with development of systemic lupus erythematosus (SLE). Here we describe a C1q-deficient patient with a compound heterozygous mutation.
Material and methods
Serum was analysed with enzyme-linked immunosorbent assay (ELISA) and Western blot for the presence of C1q, and DNA and RNA sequencing was performed to identify the mutations and confirm that these were located on different chromosomes.
Results
The medical history of the patient includes SLE diagnosis at age 11 years with cerebral involvement at age 13, various infections, osteonecrosis and hemophagocytic syndrome. Using ELISA and Western blot, we confirmed the absence of C1q in the serum of the patient. Using DNA sequencing, two mutations in the C1QC gene were identified: c.100G > A p.(Gly34Arg) and c.205C > T p.(Arg69X). With RNA sequencing we confirmed that the mutations are located on different chromosomes.
Discussion
The patient described in this case report has a compound heterozygous mutation in C1QC resulting in C1q deficiency.
Introduction
The complement system consists both of soluble and membrane-bound proteins. Activation can occur via three different pathways: the classical pathway (CP), the lectin pathway and the alternative pathway. The CP is activated when C1q is bound to immunoglobulin M (IgM), IgG immunocomplexes or pentraxins. 1 Many complement proteins are produced by hepatocytes, but cells of the immune system also produce several complement proteins. 2 Importantly, C1q is produced not by hepatocytes but largely by cells of myeloid lineage. C1q has been described to have other functions outside the complement system cascade: in the remodelling of the maternal decidua during pregnancy, during embryonic development, in neurological synapse function and in the coagulation process. 3 The C1q protein is a molecule of 480 kDa that has six identical arms. Each arm consists of three combined peptide chains: A, B and C. These peptide chains are generated from three different genes: C1QA, C1QB and C1QC, which have a synchronized transcription. 4
C1q deficiency is a rare condition with more than 70 documented cases from at least 45 different families. 5 These deficiencies all are the result of homozygous mutations in one of the three C1q genes, except in one case with a compound heterozygous mutation in C1QA. 6 Patients with C1q deficiency have various clinical presentations and outcomes. 7 Most common is the diagnosis of systemic lupus erythematosus (SLE) in early childhood and recurrent infections.6–8 Other common clinical manifestations are alopecia, Raynaud phenomenon, and involvement of the central nervous system, and Sjögren syndrome and hyper-IgM syndrome have been reported.5–7 Treatment of C1q-deficient patients mainly focuses on the treatment of symptoms. For combating the C1q deficiency itself, intravenous administration of fresh frozen plasma (FFP) is used. In rare cases allogenic hematopoietic stem cell transplantation has been performed.9,10
Here we describe a patient with C1q deficiency based on a compound heterozygous mutation in the C1QC gene. This patient was treated with FFP for more than a decade; over time it has resulted in various adverse reactions ranging from mild to anaphylactic, which led to discontinuation of the FFP therapy.
Material and methods
Patient
The patient is a 29-year-old Dutch woman diagnosed with C1q deficiency since early childhood. Blood was obtained from the patient on signing an informed consent in compliance with the Declaration of Helsinki.
Samples
Blood was collected from the patient to obtain serum as well as peripheral blood mononuclear cells (PBMCs) using Ficoll-Paque density gradient centrifugation.
Western blot
With Western blot the availability of C1q was examined by detection of the three chains of the C1q protein. Serum from the patient and normal human serum (NHS), which was used as a positive control, were applied in reduced and non-reduced sodium dodecyl sulphate conditions. Western blot was performed using previously described methods. 11
Enzyme-linked immunosorbent assay (ELISA)
C1q measurement by an in-house–developed ELISA was performed as previously described. 12 In short, plates were coated with mouse anti-human C1q ((2204), Nephrology Department, Leiden University Medical Center (LUMC)) in coating buffer (0.1 M Na2CO3, 0.1 M NaHCO3, pH 9.6), samples were incubated at 37℃ and detection was performed with rabbit anti-human C1q (Dako catalogue no. A0136) for one hour at 37℃ and subsequently a goat anti-rabbit horseradish peroxidase (Dako catalogue no. P0448), also incubated for one hour at 37℃. The substrate was added to the plates using 2,2'-azino-bis-3-ethyl benzthiazoline-6-sulphonic acid, and the signal was measured at an absorbance level of 415 nm using a Biorad iMark Microplate Absorbance Reader.
RNA isolation and complementary DNA (cDNA) preparation
PBMCs (1 × 106 cells/ml) from the patient were cultured for 72 hours in Roswell Park Memorial Institute (RPMI) (Gibco) culture medium supplemented with +1% penicillin/streptomycin, +1% GlutaMAX and +8% foetal calf serum. PBMCs were cultured either in medium alone or in the presence of dexamethasone (10 µM, Pharmacy LUMC) and interferon (IFN)-γ (200 U/ml, PeproTech), to increase C1QA/B/C expression. 13 After 72 hours RNA was isolated from the PBMCs using the mirVana microRNA isolation kit (Life Technologies catalogue no. AM1561) according to the manufacturer’s protocol. The isolated RNA was subsequently treated with DNase I, Amplification Grade (Invitrogen), and cDNA was synthesized using SuperScript III (200 U/µl, Invitrogen).
Ion Torrent sequencing
An AmpliSeq custom panel (Thermo Fisher Scientific, Waltham, MA, USA) was used to sequence the coding regions of the following genes: C4A, C2, C1S, C1R, DNASE1L3, TREX1, MASP2, C4B, C1QA, C1QB, C1QC, PLG and SERPING1. Library preparation and sequencing was performed according to manufacturer protocols on an S5 system (Thermo Fisher Scientific).
Polymerase chain reaction (PCR)
C1QC primer sequences

Sanger sequencing
PCR products from genomic DNA and cDNA were sequenced on an automated fluorescent sequencer (ABI 3730; Thermo Fisher Scientific) with the use of Big Dye Terminator (v.1.1) chemistry (Thermo Fisher Scientific). Primers used for sequencing were the same as those used for the PCR. For cDNA numbering, the A of the ATG translation initiation codon was taken as 1. This codon is codon 1. NM_172369.4 was used as a reference sequence.
Results
Patient medical history
Here we describe a Dutch woman born from two Caucasian non-consanguineous parents. She has two half brothers who are reported to be healthy. Her C1q deficiency was diagnosed at the age of 5. The patient has encountered many different clinical problems, including infections and neurological, vascular and bone complications. Her case will be presented per disease manifestation and not in chronological order. The use of FFP throughout her medical history will be discussed separately.
Infections
During the first year of her life, the patient suffered from recurring otitis and gingivitis. She was reported to be a non-responder for hepatitis B vaccination. Additionally, at the age of 6 she developed sepsis caused by Streptococcus pneumoniae. She experienced a herpes zoster infection when she was 12 years old. During adulthood, she was hospitalized with hemophagocytic lymphohistiocytosis (confirmed with a bone marrow biopsy) and pancytopenia, which was potentially induced by co-trimoxazole. During this hospitalization various infections were also diagnosed and treated accordingly: Escherichia coli and candidiasis.
Sanger sequencing
From the age of 4, clinical symptoms were compatible with SLE-like disease, with butterfly rash. At age 11 years, the diagnosis of SLE was established based on butterfly rash, oral ulcers, thrombocytopenia and positive antibodies (positive antinuclear antibodies, positive anti-Sm and positive anti-SSA). Anti-double-stranded DNA and antiphospholipid antibodies were not present. Furthermore, she experienced recurrent fevers with lymphadenopathy and vasculitis lesions of the hand and feet. Initially, the SLE was treated with hydroxychloroquine and prednisolone, with serious side effects, including weight gain (cushingoid), osteoporosis and bone infarctions. Furthermore, symptoms of fatigue, headache and arthralgia occurred during attempts to taper prednisolone treatment. On the basis of the diagnosis of SLE in combination with the earlier established C1q deficiency, and the side effects and inability to taper prednisolone, treatment with FFP was initiated.
Cerebral involvement
When the patient was 14 years old she was hospitalized with a fever, paraesthesia and difficulties with speech. Infectious causes were excluded. Magnetic resonance imaging scans of the brain were normal. Electroencephalogram showed left parieto-occipital irritative abnormalities. Liquor analysis revealed enhanced protein content without elevated cell count. With the working diagnosis of transient ischaemic attack (TIA)/partial epileptic seizure due to cerebral vasculitis, she was treated with prednisolone and carbasalate calcium. Subsequent visits at the age of 24, 25 and 28 at the Leiden University Medical Center multidisciplinary neuropsychiatric SLE (NPSLE) clinic 14 for anxiety and difficulty with speech did not reveal signs of active inflammatory NPSLE.
Vascular problems
At the age of 26 the patient developed a deep venous thrombosis, although no antiphospholipid antibodies were detected (anticardiolipin antibodies IgM/immunoglobulin G (IgG), anti-β2 glycoprotein I IgM/IgG and lupus anticoagulant). When the patient was 27 years old, she experienced a spontaneous abortion at a gestation of 8 weeks.
Bone lesions
At the age of 9 she developed an avascular necrosis of the humerus which led to a destructed right shoulder. During recent years she developed extensive bone infarctions around the knee. Because of the osteonecrosis, she underwent total hip replacement surgery at the age of 29. Osteoporosis had already been identified during childhood.
Therapy
Initially, prednisolone and hydroxychloroquine were used to treat the SLE. Because the patient was unable to taper the prednisolone and she was already diagnosed with C1q deficiency, FFP treatment was started at the age of 11 with 15 ml per kg. The FFP infusions were administered one to four times per month and were preceded by clemastine and prednisolone intravenously. CP activity was measured preceding each FFP infusion. When FFP took place each week, the CP activity was 80% to 90%; when the FFP was every two weeks, the CP activity dropped below 50%, which was in line with previous reports. 15 Anti-C1q antibodies were detectable, though not increased. There were several adverse reactions to the FFP therapy, ranging from mild urticarial to anaphylactic reaction. Despite these adverse reactions, the patient preferred the FFP therapy because of reduction of fatigue, arthralgia and number of infections. However, because of a serious anaphylactic reaction at the age of 25, FFP treatment was discontinued and her current treatment regimen consists of hydroxychloroquine, azathioprine, low-dose prednisolone, clopidogrel, bisphosphonates and cholecalciferol.
Complete absence of C1q in serum
Western blot analyses of serum from the patient and NHS (pool of four healthy adults) were analysed for the presence of C1q. The same amount of serum was applied in native, denaturing or reducing conditions. Only in the NHS lane was C1q detected (Figure 1 (a)–(c)). Therefore, we confirmed the absence of circulating C1q in the patient’s serum. Additionally, her serum was tested for C1q in ELISA format, next to 21 healthy female controls (ages 26–32 years). The C1q levels in the healthy controls had an average of 171 µg/ml C1q, whereas in the patient’s serum the C1q level was below the detection limit of 0.065 µg per ml (Figure 1(d)).
C1q protein analysis shows no C1q in the patient’s serum. Western blot analysis of serum from the patient and a control (normal human serum). The serum samples were prepared under (a) reducing condition, (b) denaturing and non-reduced condition or (c) non-reducing and non-denaturing conditions. Measurement of C1q with enzyme-linked immunosorbent assay in the serum of healthy female age-matched controls (n = 21) and (d) the patient.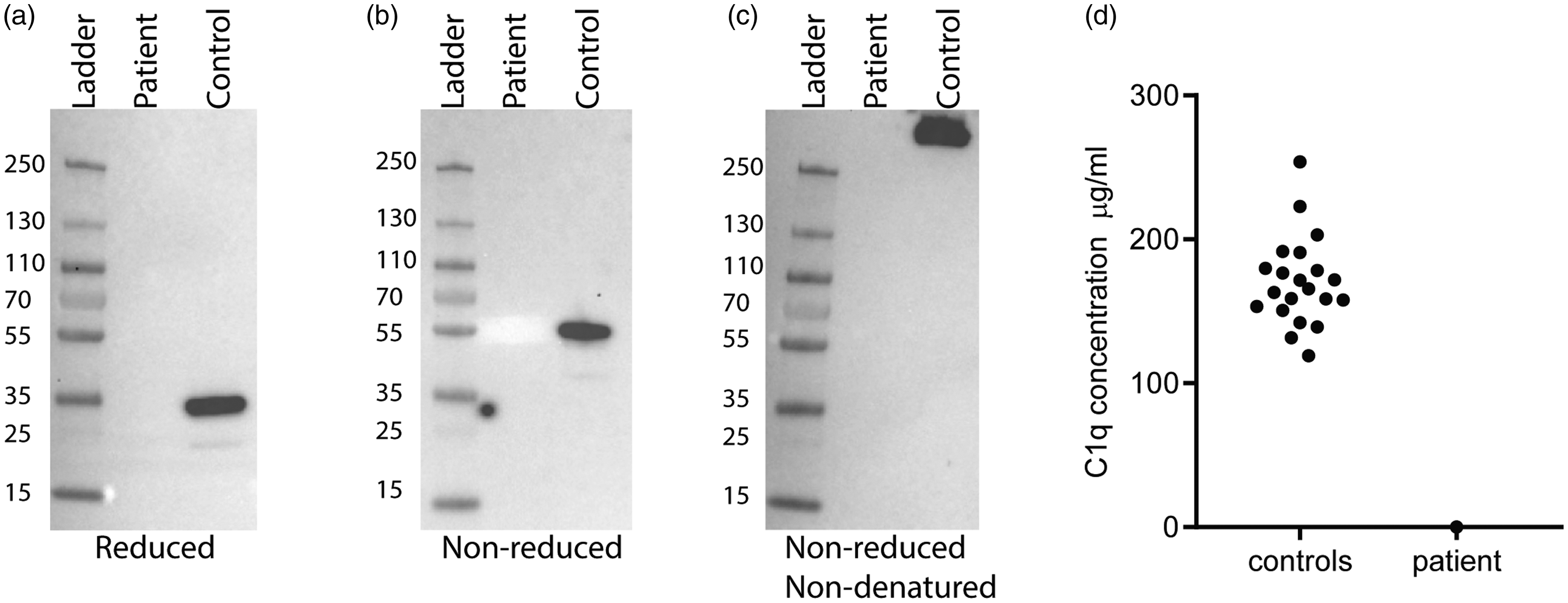
Sequencing
To determine what the mutation(s) are in this patient and where they are located, DNA and RNA sequencing was performed. Two previously described mutations were identified in the patient’s C1QC gene: c.100G > A p.(Gly34Arg) and c.205C > T p.(Arg69X). With RNA sequencing we confirmed that both mutations were heterozygous, meaning these mutations are compound heterozygous (Figure 2).
Compound heterozygous mutation located in the C1QC gene. RNA sequence analysis revealed heterozygous mutations of C1QC highlighted by the red box: c.100G > A p.(Gly34Arg) and c.205C > T p.(Arg69X).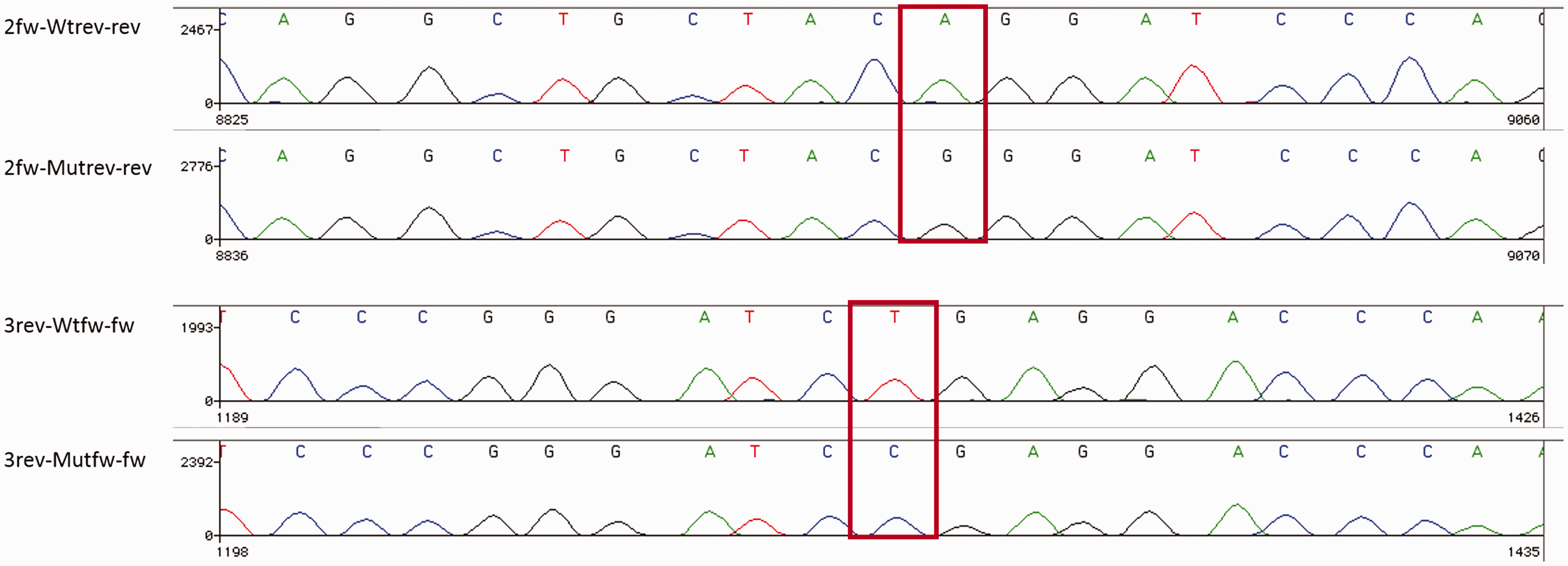
Discussion
C1q deficiency is a rare genetic disorder which is often accompanied by development of SLE. The patient described here was already diagnosed both with C1q deficiency and SLE in early childhood. She has suffered from recurrent infections, which is also a hallmark of C1q deficiency. 12 Another part of her medical history involves bone lesions. Interestingly, osteoclasts are able to produce and secrete C1q, which could be suggestive of a direct relation between C1q deficiency and development of bone lesions, although the function of C1q is unknown in this environment. 16 However, it is unclear and difficult to determine whether these bone lesions and recurrent infections are a consequence of the C1q deficiency, the SLE, the steroid treatment, or a combination of these factors. C1q is important in the clearance of apoptotic material and immune complexes. In addition to activation of the CP of the complement system, C1q has various functions independent of CP activation. 3
Previously, 15 C1q-deficient patients with NPSLE have been described in literature, with the most frequent presenting symptom being seizures (67%), which is much higher than observed in conventional SLE patients. 12 Cerebral vasculitis has also been reported in 27% of C1q-deficient NPSLE patients. The patient described here experienced neurological symptoms in adolescence and was diagnosed as having a TIA/partial epileptic seizure due to cerebral vasculitis. It is known that C1q inhibits IFN production. In patients with C1q deficiency, high IFN levels are observed in serum and cerebrospinal fluid. Recently, it was found that type I IFN stimulates microglia to engulf synaptic material, resulting in synaptic loss in the central nervous system.17,18 This could contribute to neuropsychiatric involvement in C1q deficiency.
This patient has been treated with FFP for almost 14 years. Shortly after infusion C1q levels reach their maximum and rapidly decline, whereas CP activity is sustained for a longer period of time. Even though the C1q levels and CP activity effects are relatively short lived, the symptomatic relief and substantial improvement in quality of life of the FFP treatment is sustained for several weeks. Empirically it has been established for this patient that two units of FFP every two weeks is most optimal. The FFP therapy has been accompanied by adverse events on infusion, even anaphylactoid reactions, although anti-C1q antibodies were not increased in this patient.
Of the C1q-deficient patients who have been described so far, all except one have been reported to have a homozygous mutation in the C1q genes. Here, we report the second case of C1q deficiency with a compound heterozygous mutation, in this case located in C1QC: c.100G > A p.(Gly34Arg) and c.205C > T p.(Arg69X).
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.