
Abstract
Objective
The effect of serum autoantibodies on the brain of systemic lupus erythematosus (SLE) patients remains unclear. We investigated whether serum autoantibodies, individually and assessed in groups, are associated with specific brain-MRI abnormalities or whether these structural changes are associated with other SLE-related or traditional cardiovascular disease risk factors.
Methods
All patients underwent brain 3Tesla-MRI. White matter hyperintensities (WMHs), ischemic lesions, inflammatory-like lesions and cerebral atrophy were scored. Serum autoantibodies analyzed included lupus anticoagulant (LAC), anticardiolipine (aCL) IgG and IgM (first 3 also grouped into antiphospholipid autoantibodies (aPL)), anti-dsDNA, anti-SSA, anti-SSB, anti-RNP, and anti-Sm (the latter 5 grouped into SLE-related autoantibodies). Associations were assessed using logistic regression analysis adjusted for potential confounders. Furthermore, a sensitivity analysis including anti-Beta2 glycoprotein-1 antibodies (anti-β2GP1) in the aPL group was performed and the potential modification role of the neuropsychiatric clinical status in the model was assessed.
Results
325 patients (mean age 42 years (SD 14), 89% female) were included. The following MRI-brain abnormalities were found: WMHs (71%), lacunar infarcts (21%), gliosis (11%), micro-hemorrhages (5%), large hemorrhages (2%), inflammatory-like lesions (6%) and atrophy (14%). No associations were found between individual or total SLE-related autoantibodies and inflammatory-like lesions. A higher number of positive aPL was associated with lacunar infarcts (OR 1.37 (95%CI 1.02–1.99) and gliosis (OR 2.15 (1.37–3.37)). LAC was associated with lacunar infarcts in white matter (OR 3.38 (1.32–8.68)) and atrophy (OR 2.49 (1.01–6.15)), and aCL IgG with gliosis (OR 2.71 (1.05–7.02)). Among other variables, SLE patients with hypertension presented a higher chance for WMHs (OR 5.61 (2.52–12.48)) and lacunar infarcts in WM (OR 2.52 (1.10–5.74)) and basal ganglia (OR 8.34 (2.19–31.70)), while cumulative SLE-damage was correlated with lacunar infarcts in WM (OR 1.43 (1.07–1.90)), basal ganglia (OR 1.72 (1.18–2.51)) and cerebellum (OR 1.79 (1.33–2.41)). These associations were confirmed in the sensitivity analysis.
Conclusions
Brain abnormalities in SLE represent different underlying pathogenic mechanisms. aPL are associated with ischemic brain changes in SLE, while the presence of SLE-related serum autoantibodies is not related to inflammatory-like lesions. Hypertension and cumulative SLE-damage associate with ischemic MRI-brain changes in SLE, suggesting the importance of accelerated atherosclerosis in this process.
Keywords
Introduction
Nervous system involvement in systemic lupus erythematosus (SLE) leads to a heterogeneous group of neuropsychiatric (NP) manifestations. The two main underlying pathophysiologic processes in the brain resulting in NP-SLE are thought to be inflammation and ischemia.1,2 The mechanisms that ultimately result in these pathophysiological changes and how they are related to each other remain poorly understood.
Over the past decade type I interferon (IFN) was postulated to play a central role in SLE pathogenesis by promoting feedback loops, progressively disrupting peripheral immune tolerance and driving disease activity. 3 Recent discoveries implicate IFN-alpha together with the classical complement cascade as major pathways used by microglia for synaptic pruning in mice. Chronic peripheral inflammation in SLE may play a role in the aberrant activation of microglia and subsequently stimulate synapse loss, tagging inappropriate synaptic connections between neurons and subsequently leading to cerebral dysfunction.4,5 The elevation of IFN-alpha activity has been related to autoantibody accumulation. 6 Moreover, several studies have described that autoantibody-containing immune-complexes may drive type I IFN activation.7–9 Autoantibodies may also exert a direct effect upon neurons. The disruption of the blood brain barrier (BBB) integrity may permit the influx of neuropathic antibodies, which may target synapses for engulfment by microglia. 10 Previous studies suggested that anti-dsDNA antibodies cross-react with N-methyl-D-aspartate (NMDA) receptors, and injecting these antibodies into mice causes hippocampal neuronal loss and cognitive impairment only when the BBB has been disrupted.11,12
Autoantibodies have also been associated with an ischemic pathogenic process. Antiphospholipid antibodies (aPL), especially lupus anticoagulant (LA), have been related to intracranial thrombosis. 13 The complement cascade in close relation to aPL also plays a role in microvascular injury and NP-SLE pathogenesis.14–16 Furthermore, accelerated atherosclerosis and traditional cardiovascular disease (CVD) risk factors have been involved in the ischemic process in SLE. 17
Despite the fact that imaging abnormalities are not specific for NP-SLE, magnetic resonance imaging (MRI) remains the neuroimaging technique of choice due to its superior soft tissue resolution. In a paired neuroimaging-autopsy study, Sibbit and coworkers observed that brain lesions in NP-SLE detected by MRI represent underlying cerebrovascular and parenchymal brain injury on histopathology. 18 Cerebral abnormalities that have been described in SLE on MRI are diverse; the most commonly reported is small vessel disease, especially white matter hyperintensities (WMHs) and lacunar infarcts, but also large vessel disease, inflammatory-like lesions (i.e. multifocal grey matter lesions) and brain atrophy are described.19–23 Although a fair number of studies on MRI abnormalities in SLE and NP-SLE have been published, only a few have tried to unravel the mechanisms leading to these changes. It has been proposed that focal lesions in SLE represent neuronal injury from various etiologies, ischemia and inflammation being the most important. 18 Luyendijk and coworkers described several distinct brain-MRI patterns in NP-SLE patients that were suggestive of different underlying pathogenic mechanisms. 24 The understanding of the pathogenic mechanisms leading to MRI abnormalities in SLE may be important to develop a rational prevention and treatment approach and in categorization of patients in further research.23,24
Based on this knowledge, our primary hypothesis was that the total number of SLE-related autoantibodies is associated with inflammatory-like lesions and the number of aPL autoantibodies with ischemic changes as seen on brain MRI. As a secondary objective, we analyzed if MRI abnormalities were directly related to individual autoantibodies or otherwise with other SLE-related or CVD risk factors. Overall, we aim to investigate whether the underlying immune abnormalities in SLE are associated with pathophysiological changes as seen on brain MRI.
Methods
Study population
Between September 2007 and February 2016, a total of 325 SLE patients were seen in the Leiden NP-SLE clinic and included in the present study. All patients fulfilled the American College of Rheumatology 1982 revised criteria for SLE.25,26 Our hospital is a tertiary referral centre serving as a national referral centre for NP-SLE in the Netherlands. Patients are sent by a referral rheumatologist or other medical specialist to our center when SLE patients present NP manifestations. Therefore, all patients included in this study presented NP manifestations at time of the MRI. They were admitted for a 1-day period to the Leiden University Medical Center. All patients underwent standardized multidisciplinary medical examination and extensive neuropsychological testing, serologic assessment and brain MRI. Evaluations included in the multidisciplinary assessment have been reported in detail before. 27 The attribution process of NP events to SLE and one of its underlying pathogenic mechanisms (ischemic or inflammatory) or to other etiologies was decided after multidisciplinary consensus and confirmed after re-assessment of patients at follow up as described elsewhere. 28 This study was approved by the local medical ethics committee and all patients provided written informed consent.
MRI protocol and scoring
All subjects underwent a 3 Tesla MRI in the same scanner according to a standardized protocol (Achieva; Philips Healthcare). The scanning protocol included high-resolution T1-weighted, T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences, followed by a T1-weighted sequence obtained after intravenous administration of gadolinium contrast agent. Scan parameters are shown in
Autoantibodies
Blood samples of all patients were collected from each participant at 08:00 a.m. Determinations of serum anti-dsDNA, anti-Sm, anti-RNP, anti-SSA/Ro52, anti-SSB/La and aPL including anticardiolipin (aCL), anti-Beta2 glycoprotein 1 antibodies (anti-β2GP1) and LA were performed the same day of the blood extraction in the routine clinical laboratory. IgG anti-dsDNA antibodies were detected using the Crithidia luciliae indirect immune fluorescence technique (Immuno Concepts, Sacramento, CA, USA). IgG antibodies against SS-A/Ro-52, SS-B/La, Sm, RNP, and IgG and IgM aCL and anti-β2GP1 were determined using a Phadia 250 EliA fluorescence enzyme immunoassay (Thermo Fisher Scientific, Freiburg, Germany). LA was determined using STA-Rack and STA Evolution coagulation analyzers (Stago, Parsippany, NJ, USA).
Complement levels
Levels of C3 and C4 in serum were measured using laser nephelometry. Based on the normal limits for our laboratory, C3 < 0.9 g/l and C4 < 95 mg/l were defined as low.
SLE-related activity and damage
SLE disease activity was assessed with the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K). 32 Permanent and irreversible damage due to SLE was calculated with the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index (SDI). 33 All SLEDAI-2K and SDI values were calculated without NP variables. SDI was calculated without the diabetes variable.
Cardiovascular variables
At inclusion, data on age, gender, duration of SLE, medical history and CVD risk factors (smoking, hypertension, diabetes, dyslipidemia, body mass index (BMI)) were recorded, through interviewing the patient and by studying medical records. Furthermore, at this point glucose, triglycerides, total cholesterol, low-density and high-density lipoproteins cholesterol concentrations (LDLc and HDLc) were determined. Hypertension was defined as elevated blood pressure >140/90 mmHg or receiving antihypertensive therapy. Dyslipidemia was defined according to the National Cholesterol Education Program (total cholesterol > 5.2 mmol/L, triglycerides > 1.7 mmol/L, LDLc > 3.4 mmol/L and HDLc < 1 mmol/L for men and <1.3 mmol for women) or receiving dyslipidemia therapy. 34 Cigarette smoking was divided into current and ever smoking. Diabetes was defined as a fasting plasma glucose >7.0 mmol/liter or receiving current anti-diabetic therapy. BMI was used as a continuous variable.
Statistical analysis
Demographic and clinical parameters were described as mean and standard deviations (SD) or proportions, as appropriate. The relationship between autoantibodies and MRI abnormalities was investigated through means of logistic regression analyses through which the odds ratio (OR) and 95% confidence intervals (95% CI) were calculated. First, an analysis of interaction between each of the autoantibodies (in groups and individually) and the diagnosis (NP-SLE vs. SLE without NP-SLE) on the different outcomes was conducted. If interactions were statistically significant (p < 0.1), analyses were stratified in both subgroups. If differences in the relationships (between autoantibodies and MRI abnormalities) were considered clinically relevant, all analyses were further stratified for NP-SLE diagnosis. Subsequently, analyses were conducted with groups of autoantibodies including the number of positive aPL (LA, aCL IgG and IgM) ranging from 0–3 and the number of positive SLE-related antibodies (anti-dsDNA, anti-Sm, anti-RNP, anti-SSA/Ro52, anti-SSB/La) ranging from 0–5. Afterwards, all these antibodies were included individually in separate models. Univariable regression was followed by multivariable regression. Variables from the univariable analysis with p < 0.20 were included in the multivariable model. Some variables known from the literature as potential confounders were forced into the models to test whether they confounded the main relationships of interest. Significant variables or variables with a confounding effect on the relationship between autoantibodies and MRI abnormalities were kept in the final models. Because anti-β2GP1 (IgG and IgM) were not tested in all patients, a sensitivity analysis was performed including these autoantibodies in the model as covariates first added in the aPL group (LA, aCL IgG and IgM, anti-β2GP1 IgG and IgM) ranging 0–5 and later analyzed individually; p < 0.05 was used as the level of significance. Statistical analysis was performed using SPSS version 21.0 (IBM Corp., Armonk, MY, USA).
Results
Clinical characteristics of the 325 SLE included patients
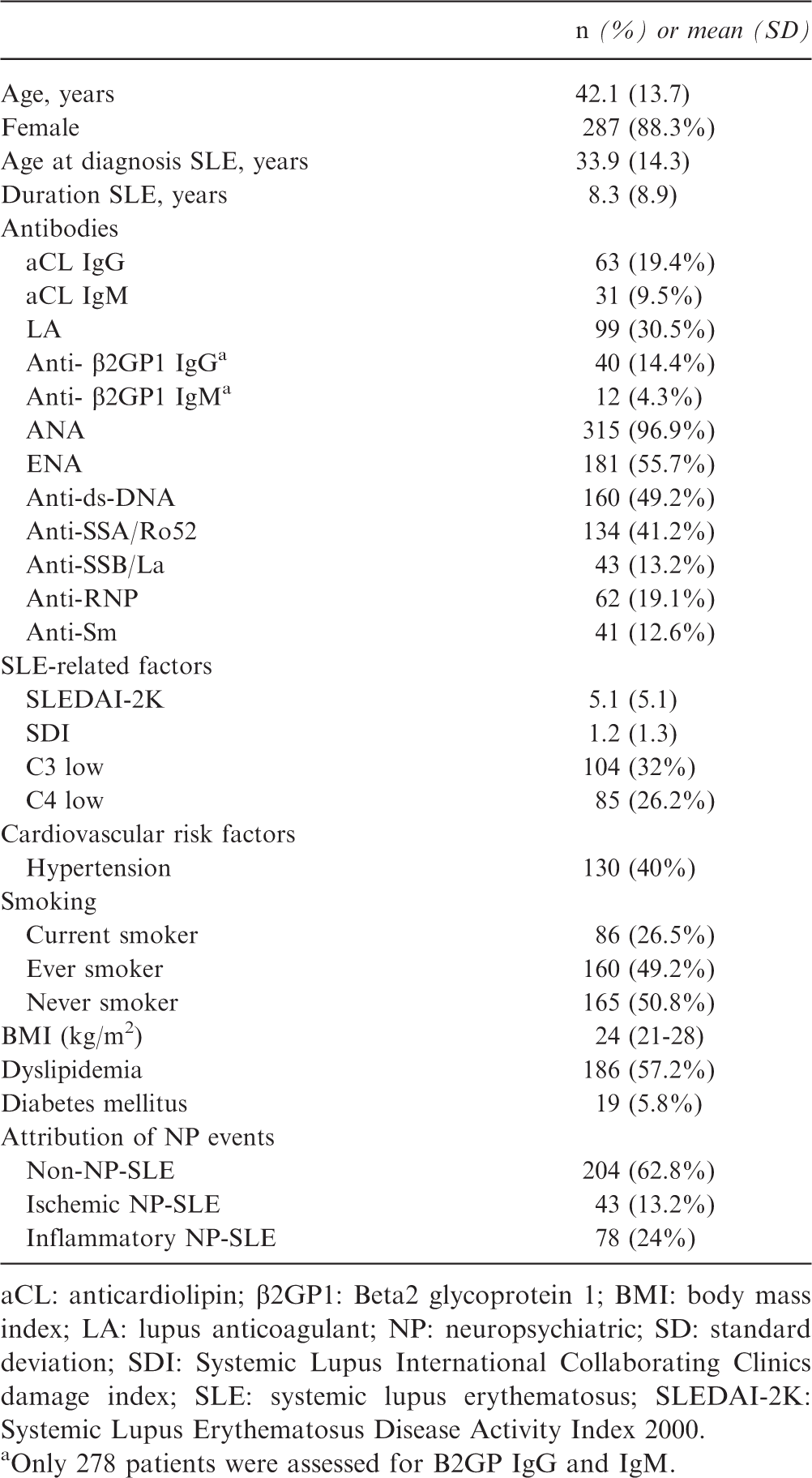
aCL: anticardiolipin; β2GP1: Beta2 glycoprotein 1; BMI: body mass index; LA: lupus anticoagulant; NP: neuropsychiatric; SD: standard deviation; SDI: Systemic Lupus International Collaborating Clinics damage index; SLE: systemic lupus erythematosus; SLEDAI-2K: Systemic Lupus Erythematosus Disease Activity Index 2000.
Only 278 patients were assessed for B2GP IgG and IgM.
Brain MRI abnormalities in SLE
Brain-MRI findings of the 325 SLE included patients
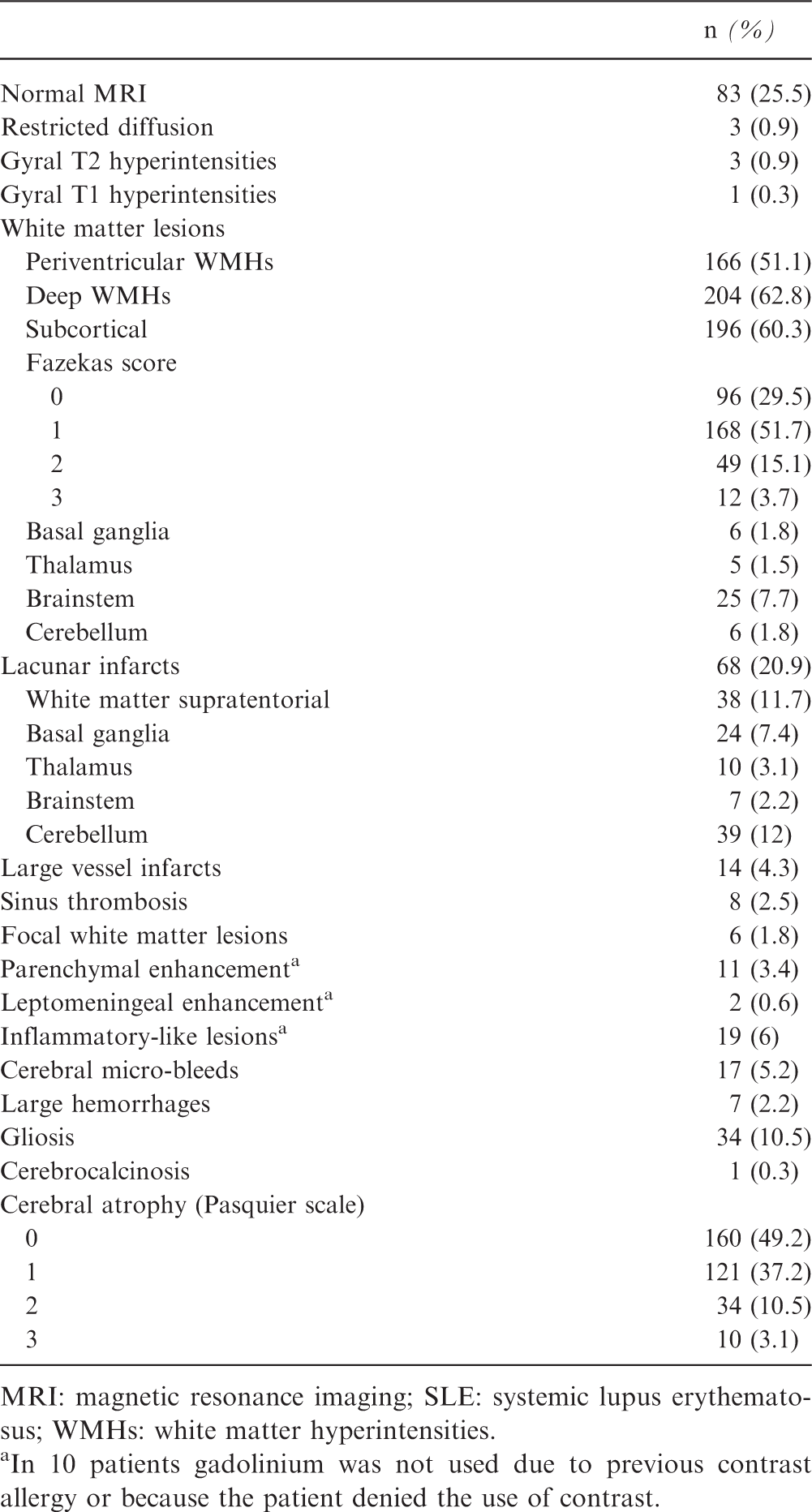
MRI: magnetic resonance imaging; SLE: systemic lupus erythematosus; WMHs: white matter hyperintensities.
In 10 patients gadolinium was not used due to previous contrast allergy or because the patient denied the use of contrast.
Relationship between groups of autoantibodies and SLE-associated MRI-brain lesions
Relationship between groups of auto-antibodies and MRI-brain lesions in 325 SLE patients
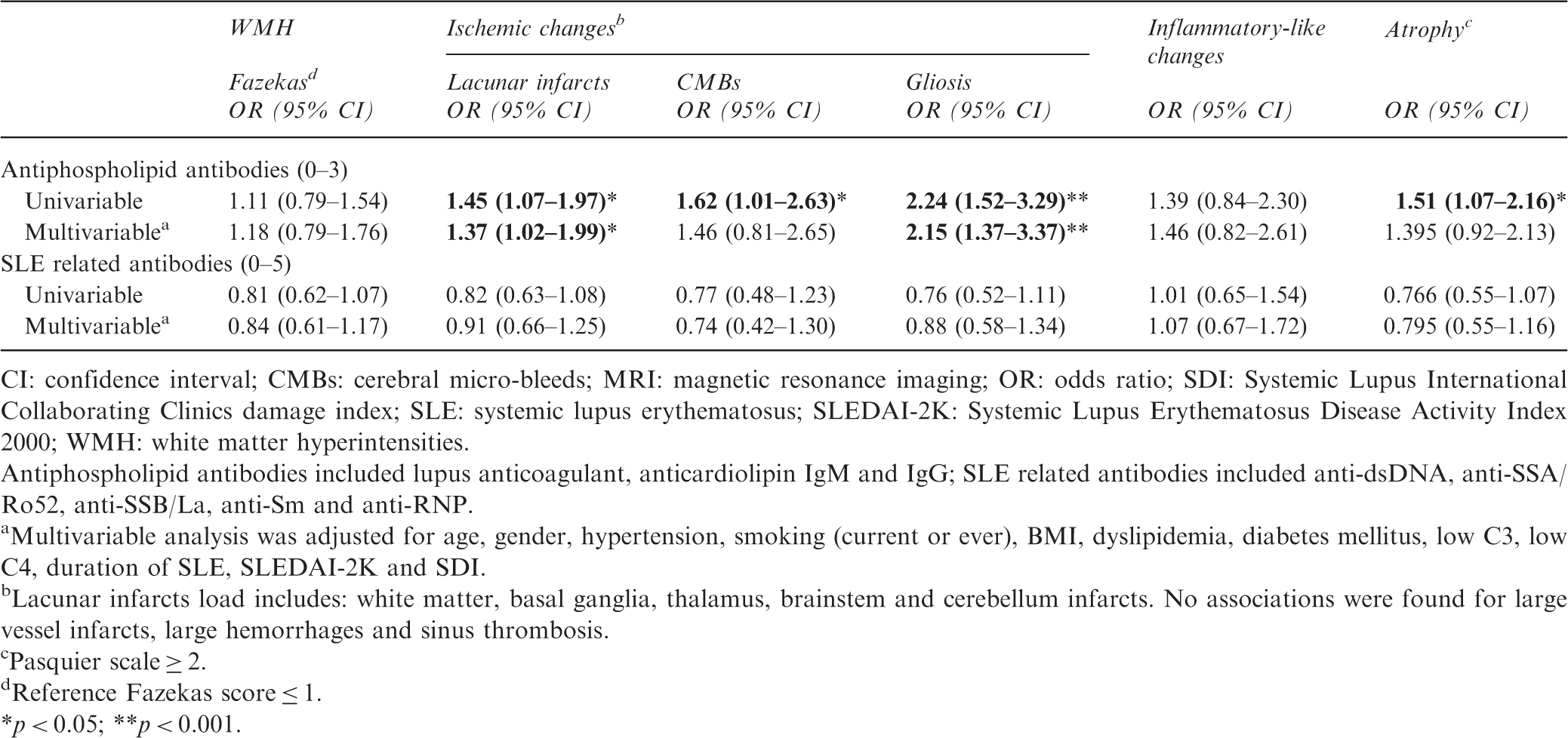
CI: confidence interval; CMBs: cerebral micro-bleeds; MRI: magnetic resonance imaging; OR: odds ratio; SDI: Systemic Lupus International Collaborating Clinics damage index; SLE: systemic lupus erythematosus; SLEDAI-2K: Systemic Lupus Erythematosus Disease Activity Index 2000; WMH: white matter hyperintensities.
Antiphospholipid antibodies included lupus anticoagulant, anticardiolipin IgM and IgG; SLE related antibodies included anti-dsDNA, anti-SSA/Ro52, anti-SSB/La, anti-Sm and anti-RNP.
Multivariable analysis was adjusted for age, gender, hypertension, smoking (current or ever), BMI, dyslipidemia, diabetes mellitus, low C3, low C4, duration of SLE, SLEDAI-2K and SDI.
Lacunar infarcts load includes: white matter, basal ganglia, thalamus, brainstem and cerebellum infarcts. No associations were found for large vessel infarcts, large hemorrhages and sinus thrombosis.
Pasquier scale ≥ 2.
Reference Fazekas score ≤ 1.
*p < 0.05; **p < 0.001.
Relationship between individual autoantibodies, CVD risk factors, SLE-specific factors, and SLE-associated MRI-brain abnormalities
Relationship between individual auto-antibodies and MRI-brain lesions in 325 SLE patients
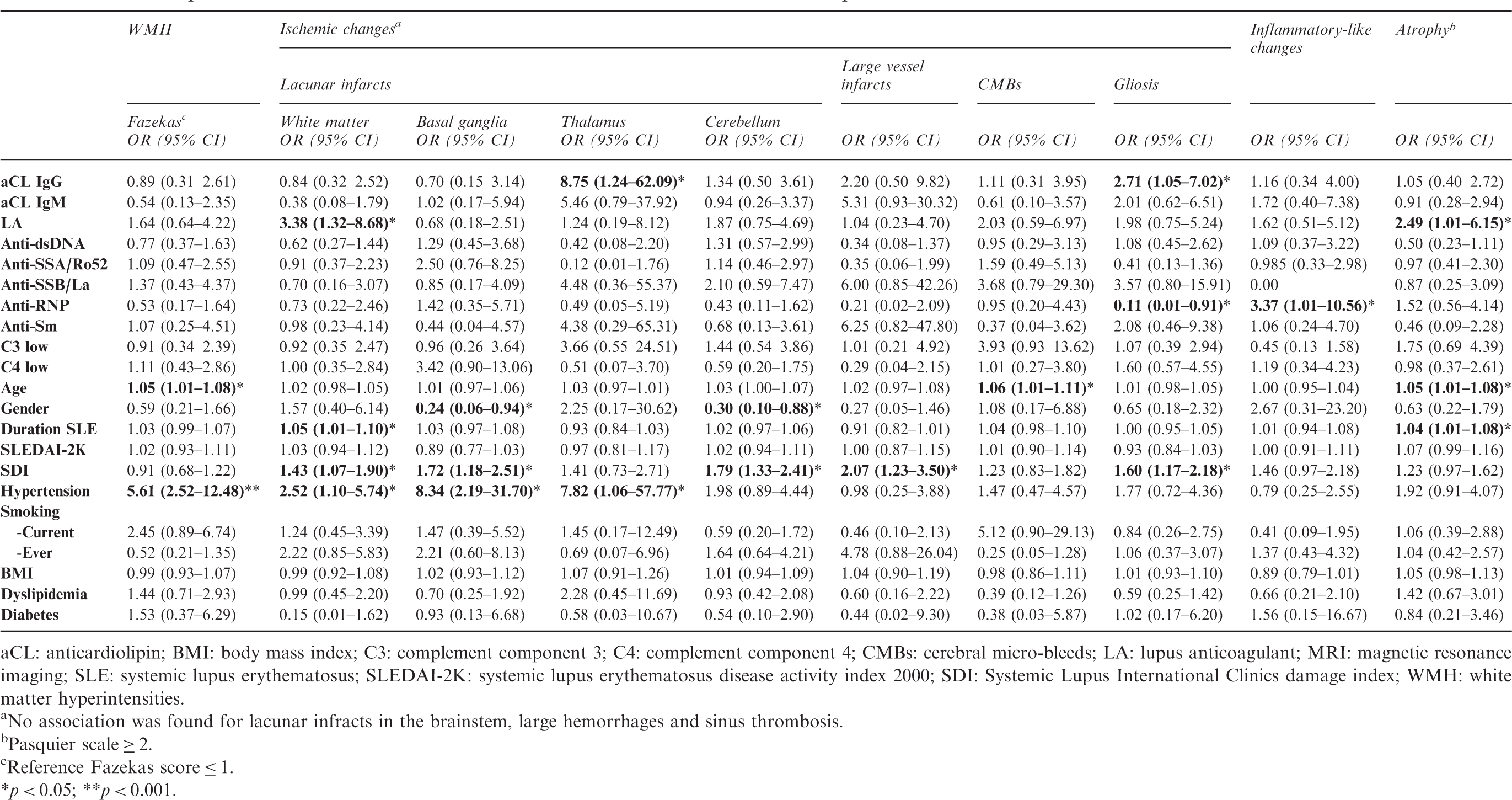
aCL: anticardiolipin; BMI: body mass index; C3: complement component 3; C4: complement component 4; CMBs: cerebral micro-bleeds; LA: lupus anticoagulant; MRI: magnetic resonance imaging; SLE: systemic lupus erythematosus; SLEDAI-2K: systemic lupus erythematosus disease activity index 2000; SDI: Systemic Lupus International Clinics damage index; WMH: white matter hyperintensities.
No association was found for lacunar infracts in the brainstem, large hemorrhages and sinus thrombosis.
Pasquier scale ≥ 2.
Reference Fazekas score ≤ 1.
*p < 0.05; **p < 0.001.
Discussion
In the present study, we have demonstrated the association of aPL, especially LA, cumulative SLE-organ damage and several CVD risk factors and the presence of ischemic changes in the brain of SLE patients. In contrast, SLE-related autoantibodies in serum, both total number and individual autoantibodies, were not associated with inflammatory-like lesions or other brain-MRI abnormalities.
Ischemic changes in the brains of SLE patients seem to be driven by aPL. Among all the serum autoantibodies analyzed, LA, but not aCL or anti-β2GP1, was associated with lacunar infarcts in the WM and with cerebral atrophy. Previous reports have also found that LA is a major risk factor for arterial thrombotic disease, especially for ischemic stroke in young women.35,36 Furthermore, a higher prevalence of MRI abnormalities, mainly lacunar and large territorial infarctions, has been found in SLE patients with antiphospholipid syndrome;21,37 LA has been suggested to play the most important role in this association. 20 The relation between LA and cerebral atrophy is more inconsistent. This relationship has been found using the Pasquier scale in a small study in SLE patients and another study including only NP-SLE patients without correction for other variables.22,38 Other studies failed to demonstrate any significant association between LA and cerebral atrophy, even when quantitative MRI-methods were used.37,39 aCL IgG was related to gliosis. We hypothesize that gliosis seen in the brain of SLE patients is driven by aCL IgG and may be part of an underlying ischemic process or due to an autoimmune-mediated glial activation.
Inflammatory-like lesions in the brain of SLE patients were not found to be related to the total number of SLE-related autoantibodies. There may be different possible explanations for these negative findings. We report a low prevalence of inflammatory-like lesions (5.8%). In the presence of a low frequency of brain-MRI inflammatory-like lesions, it is difficult to capture factors associated with it due to a lack of power. Despite this, it is difficult to interpret this frequency, as in the literature there are no other studies reporting the prevalence of inflammatory-like lesions. The MRI may have shown no abnormalities despite overt NP-SLE manifestations as it has been demonstrated with other quantitative MRI-techniques.24,40 Moreover, the inflammatory-like lesions included are still a group of heterogeneous MRI changes that may reflect different pathophysiological changes. Another reason may be the autoantibodies selected for our analysis. In the future, other serum autoantibodies (i.e. anti-ribosomal P or anti-NMDA-receptor) and autoantibodies acquired from cerebrospinal fluid (CSF) may yield stronger associations when associated with quantitative MRI techniques.
Other mechanisms, such as SLE-related and CVD risk factors, showed a correlation with brain-MRI abnormalities in SLE. Cumulative SLE organ-damage measured with SDI was found to be associated with lacunar infarcts in WM, basal ganglia and cerebellum and with cerebral atrophy. Contrary to previous reports, SDI was not related to WMHs. 41 The presence of brain MRI abnormalities in general has been previously related to higher disease severity scores and the need for more aggressive therapy. 42 Our data suggest that both infarcts and cerebral atrophy are related to chronic SLE-related damage in other organs. Therefore, patients with these brain-MRI abnormalities may have a more severe disease but also more damage due to systemic accelerated atherosclerosis affecting the whole arterial tree, which points to the importance of thrombotic/ischemic nature in leading to SLE-related damage. 43 In the general population, the presence of atherosclerosis measured by carotid intima media thickness has been related to an increased risk for brain atrophy. 44 This accelerated atherogenesis in combination with other factors such as LA and duration of disease may lead to brain infarcts and increased brain atrophy in SLE. Early diagnosis, meticulous monitoring of SLE activity, and effective use of immunosuppressive therapy may help avoid SLE-related organ damage.
Among the CVD risk factors, hypertension was correlated with a higher Fazekas score and lacunar infarcts. A relationship between long-standing hypertension and the presence of WMHs has been also described in a prospective study in a healthy population. 45 Furthermore, the strongest risk factor for ischemic stroke in the general population is hypertension. 46 Wiseman et al. recently showed an association between Fazekas score and age and hypertension in SLE patients after unadjusted univariable association. 47 Our results confirm this association after correcting for multiple confounding factors and even when the influence of the clinical status (non-NP-SLE vs. NP-SLE) was taken into account. Contrary to a previous longitudinal SLE study, 19 correlations between WMHs and aPL or SDI were not found. The different method used to assess WMHs in this previous study, semiautomatic volumetric measurements instead of Fazekas score, may explain these differences. Although hypertension and not antibodies such as aCL seem to play the most important role in the genesis of WMHs, we believe there may be other unknown SLE and non-SLE-related contributing factors leading to WMHs. Compromised BBB integrity has been recently suggested as a contributor to the pathogenesis of WMHs in the healthy population. 48 Future studies on SLE analyzing if adequate treatment of hypertension may prevent WMHs and atherosclerosis and the contribution of BBB permeability in the pathogenesis of WMHs are warranted.
Some limitations of this study should be taken into account. Due to the characteristics of our cohort and referral nuances, we were not able to measure ultrasound carotid atherosclerosis markers or calculate the cumulative dose of corticosteroids and its effect in the brain could not be investigated. Lumbar puncture is not routinely performed in the patients included in our cohort; therefore, we lack the results of CSF autoantibodies. Another limitation lies in the fact that MRIs were scored by one reader. The cross-sectional study design does not allow an interpretation of temporal or causal relationships. Another limitation may be the use of the visual Fazekas score and Pasquier scale for assessing WMHs and brain atrophy, respectively. Although these are widely used and accepted methods, the use of automated or semi-automated computer programs for quantifying lesion load and volume of WMHs and to assess measures of whole-brain atrophy may be more accurate.
Our study has important strengths. So far, most studies have focused on small groups of NP-SLE patients or on the difference between NP-SLE and SLE patients. On the contrary, we decided to include all SLE patients in the study and focus in the nature of the MRI abnormalities. We have looked into the potential modification role of the NP clinical status in the association between the investigated antibodies and the brain-MRI alterations, which was not confirmed. Furthermore, because MRI abnormalities were probably used to establish an NP-SLE diagnosis, these studies would not avoid a certain level of selection bias and circular reasoning. It is also important to consider that a proportion of patients have abnormal MRI patterns without overt clinical symptoms or a normal MRI while presenting severe NP-SLE. 42 Thus, we are convinced that including all SLE patients in the study better captures the possible underlying pathophysiological mechanisms of these MRI abnormalities. Another strength of our study is that we assessed all patients with the same high field strength (3T) using a standardized MRI protocol. To date, most studies included MRI-scans performed using different field strengths or using lower field strength (0.5–1.5T).
In summary, there is no indication that the total number or the individual SLE-related autoantibodies are associated with inflammatory-like lesions on the brain-MRI but the total number of aPL, especially the positivity for LA, are associated with ischemic brain changes, mainly with lacunar infarcts and cerebral atrophy. Furthermore, cumulative SLE-organ damage and modifiable CVD risk factors, such as hypertension, contribute to these ischemic changes, pointing to the importance of systemic accelerated atherosclerosis in SLE. We suggest that future studies should focus on CSF and other serum autoantibodies and their relationship with MRI abnormalities. Moreover, the inclusion of quantitative MRI-techniques at this point may help to understand better the underlying pathophysiological processes at a microstructural level.
Supplemental Material
Supplemental material for Are serum autoantibodies associated with brain changes in systemic lupus erythematosus? MRI data from the Leiden NP-SLE cohort
Supplemental Material for Are serum autoantibodies associated with brain changes in systemic lupus erythematosus? MRI data from the Leiden NP-SLE cohort by C. Magro-Checa, S. Kumar, S. Ramiro, L.J. Beaart-van de Voorde, J. Eikenboom, I. Ronen, J de Bresser, M.A van Buchem, T.W. Huizinga and G.M. Steup-Beekman in Lupus
Footnotes
Acknowledgements
We thank all the referral doctors and all members of the Leiden NP-SLE clinic who collected data for this study. We would like to thank Hannelore JL Beaart for her help in the database preparation.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was substantially supported by a project grant from the Dutch Arthritis Foundation (Reumafonds grant 05-1-303).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.