
Abstract
Introduction
Cuproptosis has emerged as a potential therapeutic target for colorectal cancer (CRC). This study investigated the role of ferredoxin 1 (FDX1) in regulating cuproptosis under hypoxic conditions and explored the impact of autophagy on this process in CRC.
Methods
CRC patient samples and cell lines were used in this study. Cells were exposed to hypoxia and treated with Es-Cu (a copper supplement) and rapamycin, an autophagy inducer. FDX1 expression in clinical tissues was assessed in clinical tissues using qPCR and Western blot. The CCK8 assay, EdU staining, and Transwell assay were employed to evaluate the malignant behavior of tumor cells. Copper content and DLAT oligomerization were measured. A nude mouse xenograft model was used to explore the role of FDX1 under hypoxic conditions.
Results
Compared with adjacent normal tissues, elevated FDX1 expression was observed in CRC tissues. In vitro, hypoxia or Es-Cu treatment upregulated FDX1 expression in CRC cell lines, resulting in reduced cell proliferation and increased cellular damage. FDX1 overexpression under hypoxic conditions suppressed migration, invasion, and proliferation while promoting cellular damage and DLAT oligomerization. Rapamycin-induced autophagy reversed the inhibitory effects of FDX1 overexpression on CRC cells. In vivo, rapamycin treatment attenuated the tumor-suppressive effects of FDX1 overexpression in nude mouse xenograft models.
Discussion
This study demonstrated that hypoxia-induced autophagy inhibits FDX1-mediated cuproptosis, leading to resistance to copper-induced cell death in CRC cells. Targeting the autophagy pathway may provide a novel therapeutic strategy to overcome resistance to cuproptosis and improving CRC treatment outcomes.

Introduction
In 2020, colorectal cancer (CRC) ranked third among all diagnosed malignant tumors worldwide. 1 In China, the incidence of CRC has also increased rapidly. Compared to 1990, the number of CRC cases and deaths in China had risen by 474.03% and 230.14%, respectively, in 2019. 2 Despite advancements in surgical techniques and adjuvant chemotherapy/radiotherapy improving the survival rates of CRC patients, the prognosis for advanced CRC remains poor. Factors contributing to this poor prognosis include tumor recurrence, metastasis, and treatment resistance.3–5 Further research into the mechanisms of tumorigenesis and progression in CRC, as well as the identification of potential biological targets associated with tumor recurrence and metastasis, is crucial for improving patient prognosis and developing more effective clinical treatment strategies.
Copper ions play a significant role in tumor development. However, excessive accumulation of copper ions in cells can trigger a copper-dependent cell death process known as cuproptosis. Cuproptosis, a novel form of cell death, was first described by Tsvetkov et al. in 2022. Cuproptosis is defined as a copper-dependent cell death caused by the direct binding of copper ions to lipidated components in the tricarboxylic acid (TCA) cycle during mitochondrial respiration. 6 This binding leads to the aggregation of lipidated proteins and the subsequent downregulation of iron-sulfur cluster proteins, inducing proteotoxic stress and ultimately resulting in cell death. 6
Ferredoxin 1 (FDX1) is a key regulatory gene in cuproptosis, functioning as a critical factor in transferring electrons from NADPH to mitochondrial cytochrome P450, converting Cu2+ to the more toxic Cu+ 6. A previous study has confirmed that Elesclomol directly target the FDX1 gene. FDX1 is involved in ATP production, participating in glucose metabolism, fatty acid oxidation, and amino acid metabolism. 7 Hepatocellular carcinoma patients with high FDX1 expression exhibited significantly prolonged survival compared with those with low FDX1 expression. 8 The anti-tumor role of FDX1 in clear cell renal cell carcinoma was further confirmed by Wang et al. 9 In colorectal cancer, FDX1 expression is positively correlated with the prognosis of patients with rectal adenocarcinoma. 10 Specifically, FDX1 expression participates in the cuproptosis process, enhancing the antitumor effect of 4-Octyl itaconate. 11 However, the mechanisms underlying FDX1 in CRC warrant further exploration.
Tumor hypoxia, a common feature of the tumor microenvironment caused by inadequate blood supply, is a hallmark of tumors. Under hypoxic conditions, tumor cells adapt to this unfavorable environment by upregulating autophagy. 12 Autophagy activation facilitates tumor cells recycling damaged organelles and proteins, and providing survival advantages under hypoxia or nutrient-deficient conditions by maintaining cellular homeostasis. 13 This phenomenon has been extensively documented in CRC.14–16
This study hypothesizes that colorectal cancer cells employ enhanced autophagy mechanisms under hypoxic conditions to evade cuproptosis induced by copper ion overload. Specifically, hypoxia may induce autophagy that mitigate the toxicity of copper ions within cells, thereby enhancing the survival of colorectal cancer cells. The aim of this study is to investigate the role of autophagy in colorectal cancer cells under hypoxic conditions, elucidate the regulatory role of autophagy in FDX1-mediated cuproptosis, and identify potential therapeutic targets for the colorectal cancer treatment.
Methods
Clinical sample collection and detection
Patients diagnosed for the first time with colorectal cancer at Nan Chong Central Hospital were included in this study. Informed consent were obtained from patients before surgery and this study strictly adhered to the Declaration of Helsinki. Clinical tissues collected from patients during surgery were stored in liquid nitrogen or fixed in 4% paraformaldehyde. Detailed clinical characteristics of the patients were recorded for subsequent analysis. This study was approved by the Ethics Committee of Nan Chong Central Hospital (approval No.: 2024-087).
Cell culture, hypoxia, and treatment
Cell lines, including SW480, SW620, HCT116, LoVo, RKO, and the normal cell line FHC, were maintained in DMEM (ServiceBio, Wuhan, China), supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin and cultured at 37°C, 100% humidity, and 5% CO2. RKO and LoVo cells were selected for subsequent experiments. Cells cultured under normoxic conditions were transferred to a hypoxic chamber with 2% oxygen while other parameters remained unchanged. After 24 h of culture, a hypoxia model was established. The human colorectal cancer cell lines used in this study were purchased from the China Center for Type Culture Collection (CCTCC). Prior to the experiments, the cell lines were authenticated through STR profiling and tested for mycoplasma contamination.
qPCR
Primer sequences for real time PCR in this study.

TTCAACCTGTCACCTCATCTTTG (5′-3′, forward primer of FDX1) and TGCCAGATCGAGCATGTCATT (5′-3′, reverse primer of FDX1), CATGTACGTTGCTATCCAGGC (5′-3′, forward primer of ACTB) and CTCCTTAATGTCACGCACGAT (5′-3′, reverse primer of ACTB).
Cell proliferation activity detection
CCK-8 (Cell counting kit-8)
Cell viability was assessed using the Cell Counting Kit-8 obtained from Beyotime (C0041, Shanghai, China). Cells were seeded into 96-well plates after treatment (hypoxia, Es-Cu treatment, and/or transfection), and cell viability was measured after 24 h of culture. Briefly, CCK-8 working reagent was added to each well, followed by incubation for 1 h. Finally, the cell supernatant was collected, and the absorbance at 450 nm (OD450 nm) was measured to calculate cell survival based on a standard curve.
EdU staining
Cell proliferation was assessed using the Enhanced Cell Counting Kit-8 (C0075 L, Beyotime, Shanghai, China). Following treatment, cells were incubated with 50 μM EdU medium (100 μL per well) for 2 h. After discarding the medium, the EdU Click reaction solution was added, and cells were observed under a fluorescence microscope. Red fluorescence indicated EdU-positive signals, and blue fluorescence indicated cell nuclei.
Autophagy detection
Western blot
Cells (1 × 106 cells) were lysed, and total protein was extracted after centrifugation. Protein concentration was quantified using the BCA kit (Beyotime, Shanghai, China). Equal amounts of protein (20 μg per lane) were separated by SDS-PAGE and transferred to PVDF membranes. Subsequently, the PVDF membranes were incubated with primary antibodies and secondary antibodies, followed by exposure to X-ray film. The primary antibodies used included LC3B (BM4827, 1:2000; Boster, Shanghai, China), Beclin 1 (PB9076, 1:2000; Boster, Shanghai, China), P62 (BA2849, 1:2000; Boster, Shanghai, China), FDX1 (M05441, 1:1500; Boster, Shanghai, China), HIF-1α (GB111339, 1:1000; ServiceBio, Wuhan, China), and DLAT (A04469-2, 1:1500; Boster, Shanghai, China). β-Actin was used as an internal control.17,18 Band intensities were analyzed with ImageJ software, and target protein levels were expressed as the ratio of target protein intensity to β-actin intensity. Three independent biological replicates were performed for each experiment, and representative blots are shown in the figures.
Immunofluorescence
Tissues were fixed, embedded in paraffin, and sectioned (4 μm). Tissue sections were deparaffinized and incubated with the LC3B antibody (BM4827, 1:200; Boster, Shanghai, China) and secondary antibodies. LC3B expression and localization in tissues were visualized using a fluorescence microscope.
Detection of copper-induced cell death
Copper content detection
Human colorectal cancer tissues and adjacent non-cancerous tissues were homogenized and centrifuged. The tissue supernatant was collected after homogenization, and the copper content was quantified using a Copper Assay Kit (MAK127, Sigma, USA) was used to quantified the copper content according to the manufacturer’s instructions.
Flow cytometry
To detect the oxidative stress of cells, ROS was measured. Briefly, the DCDH-DA probe was diluted in serum-free medium (1:1000) and then the probes were loaded into cells via cell culture medium. Cells were incubated at 37°C in the dark for 30 min, and then washed twice with serum-free medium. Cells were transferred to flow cytometry tubes for analysis, and the data were analyzed using FlowJo software.
Transwell assay
Cells were seeded in the upper chamber of Transwell inserts (serum-free medium), while the lower chamber contained medium supplemented with 10% fetal bovine serum. After 24 h of incubation, the chambers were fixed in 4% paraformaldehyde, and stained with crystal violet. Migrated or invaded cells were counted under a microscope to assess cell migration or invasion ability. For invasion assays, Matrigel was coated in the upper chamber prior to cell seeding. Five fields were randomly selected per group to evaluate migration and invasion abilities.
Nude mouse xenograft model
A total of 60 BALB/c-nu/nu mice (Six-week-old, ∼20 g) were housed in an SPF-grade animal facility, under controlled conditions (22 ± 2°C), humidity (∼60%), and a 12-h light/dark cycle. Mice were provided with sufficient food and water throughout the experiment. LoVo cells (1 × 106 cells per animal) were subcutaneously injected into the axillary region (N = 6 per group). These mice were divided into groups (N = 6 per group), with half of the total animals being housed under standard (normoxic, 21% O2) conditions and the other half housed in a hypoxic chamber (10% O2) for the duration of the experiment. Tumor length and width were measured every 5 days, and tumor volume (V = a*b*b/2) was calculated. The mice were rapidly euthanized by cervical dislocation on the 28th day of the experiment. Tumor masses were measured and then preserved in liquid nitrogen or fixed in 4% paraformaldehyde. Ethical approval for the animal experiments was obtained from the Laboratory Animal Care and Use Committee of the Institute of Biological and Medical Engineering, Guangdong Academy of Sciences (Approval No. K224-01-186-491). The animal study in this research strictly adhered to the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
Histological analysis
H&E staining
Tissues from animals or patients were fixed in 4% paraformaldehyde for 24 h, dehydrated through a graded alcohol series, and embedded in paraffin. Tissue sections were deparaffinized and stained with Harris hematoxylin for 3–8 min, differentiated with 1% hydrochloric acid alcohol, and counterstained with eosin for 1–3 min. Slides were dehydrated, mounted, and examined under a microscope.
Immunohistochemistry
Paraffin-embedded sections were deparaffinized and subjected to antigen retrieval in EDTA buffer (pH 9.0) using a microwave oven. Endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxide. Sections were incubated with 3% BSA and then with the Ki67 primary antibody (GB111141, 1:500; ServiceBio, Wuhan, China) at 4°C overnight. Subsequently the sections were incubated with secondary antibody (HRP-conjugated) for 50 min at room temperature. Sections were developed with DAB solution and counterstained with hematoxylin. Finally, histopathological examination was performed using light microscopy.
TUNEL assay
Paraffin-embedded sections were incubated with the TUNEL reaction mixture for 1 h at 37°C, mounted with an antifade solution, and examined under a fluorescence microscope. The average number of TUNEL-positive cells was quantified and analyzed using ImageJ software.
Data analysis
Data were presented as mean ± SD in this study. Statistical analyses were performed using SPSS 22.0 (IBM, USA). Comparisons between two groups were performed using Student’s t-test, while data involving more than two groups were analyzed using one-way ANOVA with Tukey tests. All experiments included three independent biological replicates in this study, and a P-value <0.05 was considered as statistically significant.
Results
Characteristics of FDX1 in colorectal cancer
To determine the expression characteristics of FDX1 in colorectal cancer, immunohistochemistry was performed to detect FDX1 expression in clinical tissues from colorectal cancer patients. The results showed that FDX1 was highly expressed in tumor (T) regions and lowly expressed in adjacent normal (PT) regions (Figure 1(a)). Western blot analysis confirmed the IHC results, indicating upregulated FDX1 expression in tumor tissues along with increased HIF-1α expression (Figure 1(b)). The copper content in tumor tissues was significantly elevated (Figure 1(c)), and increased with tumor progression in colorectal cancer patients (Figure 1(d)). According to the GEPIA database, FDX1 expression was upregulated in gastrointestinal tumors, including colon cancer, esophageal squamous cell carcinoma, rectal cancer, gastric cancer, and pancreatic cancer (Figure 1(e)). The prognostic significance of FDX1 expression was evaluated using Kaplan-Meier Plotter database. The results showed that a correlation between elevated FDX1 levels and poorer survival outcomes reduced overall survival (OS), indicating that high FDX1 expression was related to poorer survival rates (Figure 1(f)). FDX1 expression is upregulated in colorectal cancer tissues and correlates with poor prognosis. (a) Immunohistochemical staining of FDX1 in colorectal cancer tissues (T) and adjacent normal tissues (N). (b) Western blot analysis of FDX1 and HIF-1α expression in colorectal cancer tissues (T) and adjacent normal tissues (N). (c) Quantification of copper content in colorectal cancer tissues and adjacent normal tissues. (d) Quantification of copper content in colorectal cancer tissues at different stages. (e) FDX1 expression in various gastrointestinal tumors based on the GEPIA database. (f) Kaplan-Meier analysis of the association between FDX1 mRNA expression and overall survival in colorectal cancer patients. *p < .05, **p < .01.
Potential mechanism of FDX1 in colorectal cancer
To explore the role of FDX1 in colorectal cancer, qPCR was first used to measure FDX1 expression levels in colorectal cancer cell lines. As shown in Figure 2(a), FDX1 expression was low in colorectal cancer cell lines under normoxic conditions; however, hypoxia or Es-Cu treatment significantly upregulated FDX1 expression (Figure 2(b)). CCK-8 assays showed that both hypoxia and Es-Cu treatment significantly reduced cell proliferation in LoVo and RKO cell lines, with the most pronounced suppression observed under combined hypoxia and Es-Cu treatment (Figure 2(c)). TUNEL assays indicated that both hypoxia and Es-Cu treatment induced significant cell damage, with the most severe damage observed under combined treatment (Figure 2(d)). Hypoxia and Es-Cu treatment upregulate FDX1 expression and inhibit proliferation and induce cell damage in colorectal cancer cells. (a) qPCR analysis of FDX1 expression in colorectal cancer cell lines under normoxic conditions. (b) Western blotting of FDX1 and HIF-1αin LoVo and RKO cells. (c) CCK8 assay of cell proliferation in LoVo and RKO cells. (d) TUNEL assay of cell damage in LoVo and RKO cells. *p < .05, **p < .01.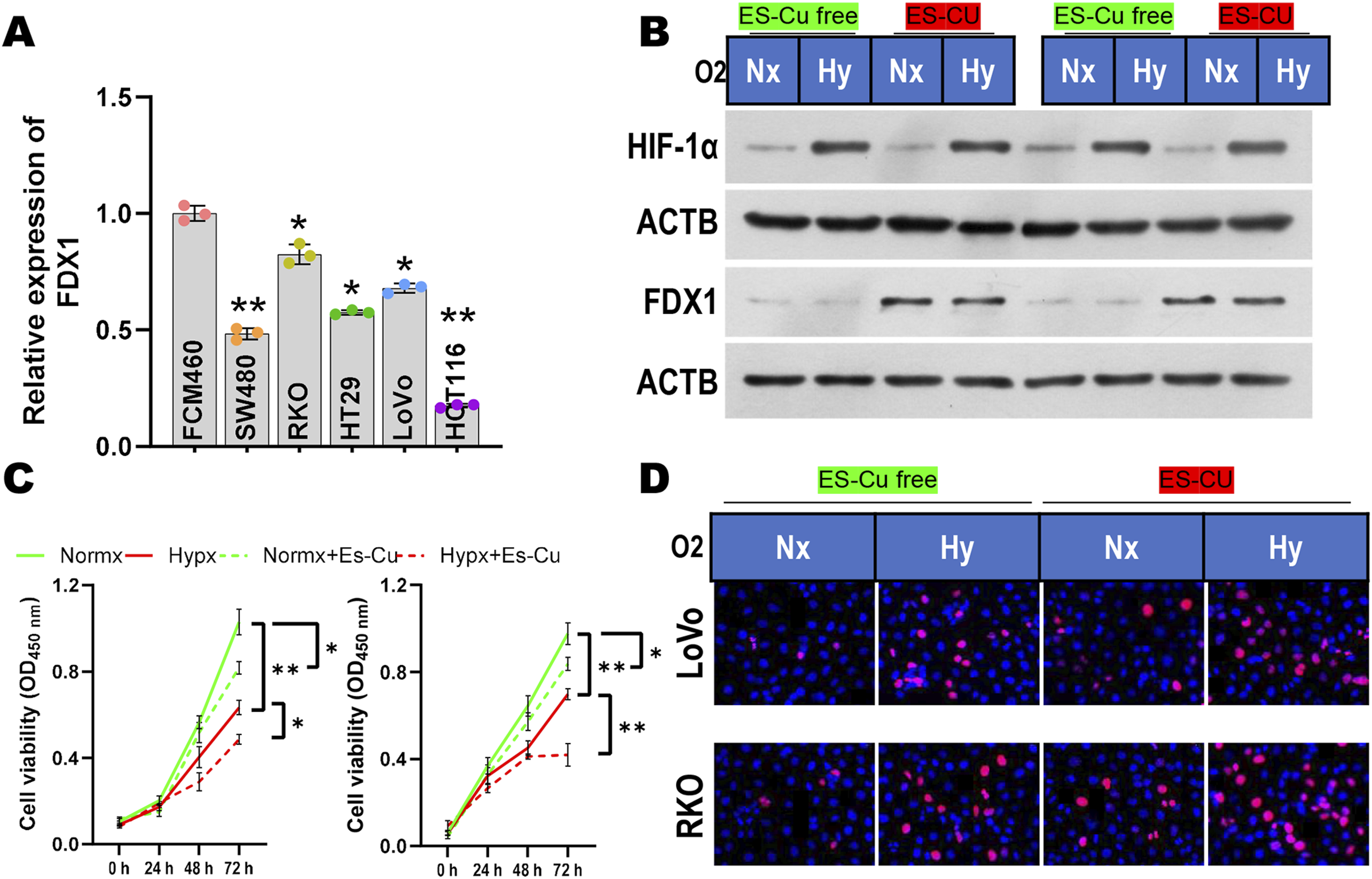
Regulation of colorectal cancer by FDX1 under hypoxic conditions
To explore the regulatory effects of FDX1 on colorectal cancer under hypoxic conditions, LoVo and RKO cells were treated with Es-Cu and exposed to hypoxia in LoVo and RKO cells, with FDX1 expression either silenced or overexpressed. As shown in Figure 3, hypoxic conditions significantly inhibited cell migration compared to normoxic conditions. Hypoxia also increased increased cell damage and reduced proliferation (Figure 3(a), 3(b), 3(c)). Compared with the control group, FDX1 silencing enhanced cell migration and proliferation while reducing cell damage (Figure 3(a), 3(b), 3(c)). Conversely, FDX1 overexpression reduced migration, invasion, proliferation, while increasing cell damage in colorectal cancer cells (Figure 3(a), 3(b), 3(c)). Western blot analysis revealed that FDX1 overexpression increased DLAT oligomerization in colorectal cancer cells, whereas silencing FDX1 decreased DLAT oligomerization levels (Figure 3(d)). Additionally, both Western blot and immunofluorescence results indicated significantly enhanced autophagy under hypoxic conditions, as evidenced by upregulated LC3B II and Beclin 1 expression and downregulated P62 expression (Figure 4(a), 4(b)). FDX1 overexpression inhibits the malignant behavior of colorectal cancer cells under hypoxic conditions. (a) Transwell assay of cell migration and invasion in LoVo and RKO cells with FDX1 silencing or overexpression under hypoxic conditions. (b) EdU staining of cell proliferation in LoVo and RKO cells. (c) TUNEL assay of cell damage in LoVo and RKO cells. (d) Western blot analysis of DLAT oligomerization in LoVo and RKO cells. *p < .05, **p < .01. Hypoxia induces autophagy in colorectal cancer cells. (a) Western blot analysis of LC3B, Beclin 1, and P62 expression in LoVo and RKO cells under hypoxic conditions. (b) Immunofluorescence staining of LC3B in LoVo and RKO cells under hypoxic conditions. *p < .05, **p < .01.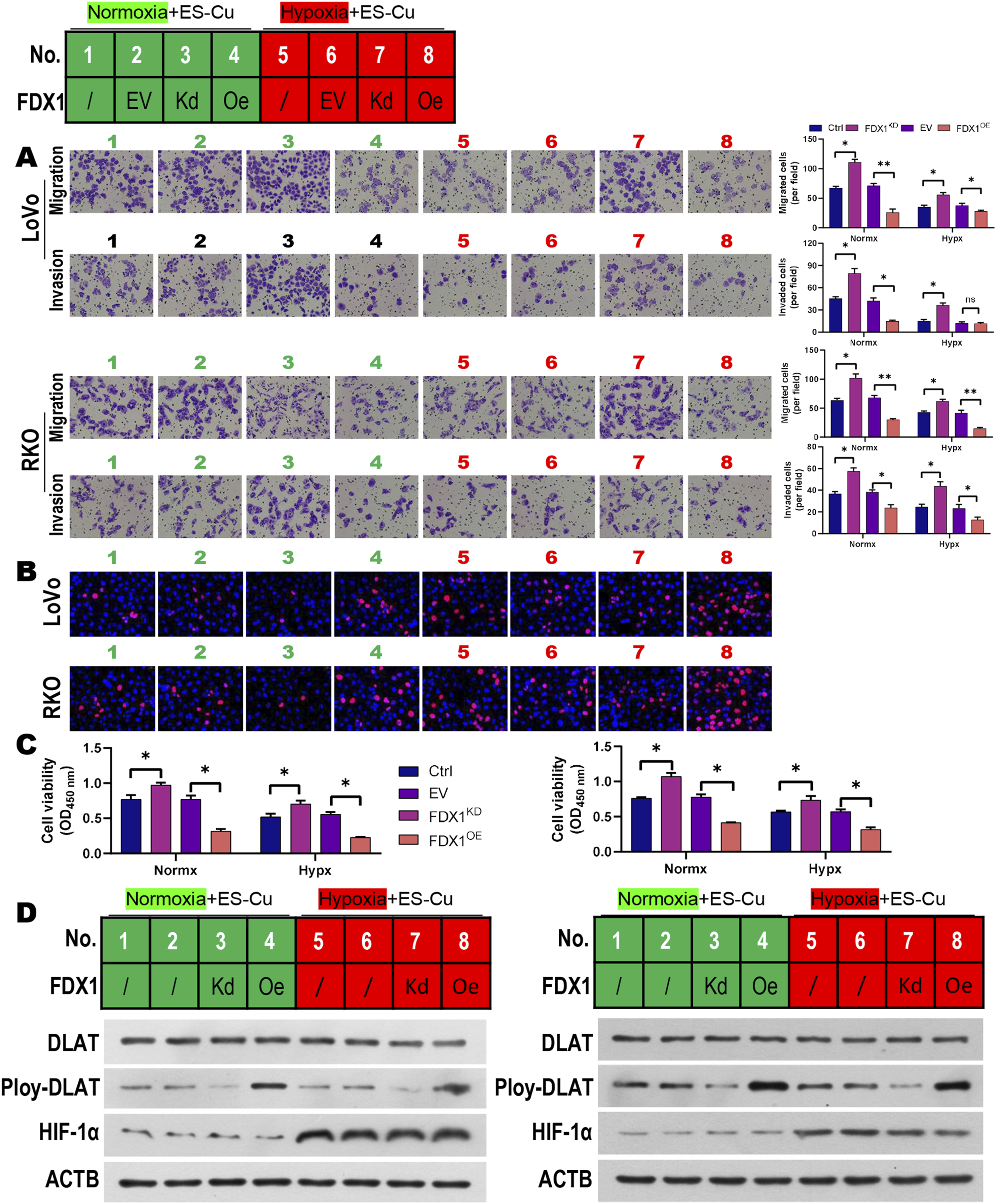

Regulation of hypoxia-induced autophagy on FDX1-Mediated cuproptosis in colorectal cancer
To investigate the impact of hypoxia-induced autophagy on FDX1-mediated cuproptosis in colorectal cancer cells, cells were exposed to hypoxia, treated with Es-Cu, overexpressed with FDX1, and further treated with rapamycin (an autophagy activator). As shown in Figure 5, rapamycin treatment significantly enhanced autophagy in colorectal cancer cell lines (Figure 5(a), 5(b)). Rapamycin reversed the suppressive effects of FDX1 overexpression on migration, invasion, and cell viability, as well as the increase in cell damage in colorectal cancer cells (Figure 5(c), 5(d), 5(e)). However, Western blot results showed no significant changes in DLAT oligomerization levels in colorectal cancer cells following rapamycin treatment (Figure 5(e)). Hypoxia-induced autophagy attenuates FDX1-mediated cuproptosis in colorectal cancer cells. (a) Western blot analysis of LC3B, Beclin 1, and P62 expression in LoVo and RKO cells treated with rapamycin under hypoxic conditions. (b) Immunofluorescence staining of LC3B in LoVo and RKO cells. (c) Transwell assay of cell migration and invasion in LoVo and RKO cells. (d) EdU staining and TUNEL assay of cell proliferation and damage in LoVo and RKO cells. (e) Western blot analysis of DLAT oligomerization in LoVo and RKO cells. *p < .05, **p < .01.
Effects of autophagy on FDX1-Mediated cuproptosis on tumor growth
To clarify the effects of autophagy on FDX1-mediated cuproptosis on tumor growth in vivo, LoVo cells were implanted into nude mice, housed in a hypoxic chamber. As shown in Figure 6, tumor growth was significantly reduced under hypoxic conditions compared with normoxic conditions. Regardless of oxygen conditions, FDX1 overexpression resulted in reduced tumor growth and tumor weight significantly. However, rapamycin treatment in nude mice implanted with FDX1-overexpressing LoVo cells restored tumor growth (Figure 6(a), 6(b), 6(c), 6(d)). Additionally, FDX1 overexpression increased necrotic areas in tumor tissues and TUNEL-positive regions, whereas rapamycin treatment significantly reduced necrosis and TUNEL-positive areas (Figure 6(e), 6(f)). qPCR and Western blot analysis confirmed that FDX1 expression was upregulated under hypoxic conditions, and FDX1 overexpression was associated with increased DLAT oligomerization (Figure 6(g), 6(h)). Finally, Western blot analysis of tumor tissues indicated enhanced LC3B and Beclin 1 expression under hypoxic conditions, and further autophagy induction following rapamycin treatment (Figure 6(i)). Autophagy inhibition enhances the tumor-suppressive effects of FDX1 overexpression in vivo. (a) Representative images of tumors from nude mice implanted with LoVo cells under different conditions. (b, c, d) Tumor volume (b, c) and weight of nude mice implanted with LoVo cells under different conditions. (e) H&E staining and immunohistochemical staining of Ki67 in tumor tissues from nude mice implanted with LoVo cells under different conditions. (f) TUNEL assay of cell damage in tumor tissues from nude mice implanted with LoVo cells under different conditions. (g) qPCR analysis of FDX1 expression in tumor tissues from nude mice implanted with LoVo cells under different conditions. (g) Western blot analysis of FDX1 and DLAT oligomerization in tumor tissues. (h) Western blotting of LC3B and Beclin 1 expression in tumor tissues from nude mice implanted with LoVo cells under different conditions. *p < .05, **p < .01, ns noted no significance.
Discussion
As China transitions into an aging society, the burden of colorectal cancer is increasing, significantly impacting survival and quality of life. In-depth research into the mechanisms of colorectal cancer and the development of more effective therapeutic strategies are essential for disease control. This study explored the effects of FDX1-mediated cuproptosis mechanisms on the malignant behavior of colorectal cancer under hypoxic conditions. This study is the first to propose that hypoxia-induced autophagy in colorectal cancer confers tumor cell tolerance to cuproptosis. This finding explains the upregulation of FDX1 expression in colorectal cancer and enhances our understanding of the mechanisms underlying colorectal cancer initiation and progression.
As a key regulator in cuproptosis, FDX1 has garnered increasing attention in recent years. Studies have shown that FDX1 expression is upregulated in gastric cancer. 19 Knockdown of FDX1 in gastric cancer cells inhibits malignant behaviors. 20 Similarly, Sun et al. 21 have found that FDX1 is highly expressed in gastric cancer; however, after inhibiting cuproptosis, significantly enhances cell survival. In colorectal cancer, FDX1 expression is downregulated, overexpression of FDX1 exerts tumor-suppressive effects by inhibiting EMT both in vivo and in vitro experiments. 22 Similarly, compared with adjacent non-cancerous tissues or normal tissues, FDX1 expression is downregulated in rectal cancer tissues and is significantly associated with patient prognosis. 11 In this study, however, we found that FDX1 expression was upregulated in colorectal cancer tissues, accompanied by significantly elevated copper ion levels. These conflicting findings may be attributed to the presence of regulatory mechanisms in tumor cells to enhance tolerance to cuproptosis. Therefore, we propose that the upregulation of FDX1 in colorectal cancer tissues may reflect tumor cell adaptation to FDX1-mediated cuproptosis.
Solid tumors frequently develop a hypoxic microenvironment. Under hypoxic conditions, tumors enhance their survival by altering metabolic pathways and activating adaptive mechanisms. 23 Hypoxia induces autophagy, which promotes tumor cell survival under stress through the degradation and recycling of cellular components. 24 Tumor cells widely employ this survival strategy to sustain proliferation and resist adverse conditions. For instance, hypoxia-induced autophagy in colorectal cancer cells accelerates tumor EMT. 25 Additionally, hypoxia upregulate HIF-1α expression, inducing autophagy and promoting cisplatin resistance in bladder cancer. 26 Therefore, hypoxia-induced autophagy may explain the observed tolerance to cuproptosis in colorectal cancer. Indeed, copper ions play a critical role in cuproptosis. Studies have shown that copper ions induce autophagy in breast cancer.27,28 In polycystic ovary syndrome (PCOS), FDX1 regulates autophagy-related proteins, ATG3 and ATG7, thereby modulating the proliferation and apoptosis of KGN cells. 29 Copper ions induce autophagy by binding to ULK1 and ULK2, promoting lung cancer progression. 30 However, study in non-small cell lung cancer has shown that inhibiting autophagy through ATG5 siRNA or 3-MA significantly increases DSF/Cu-induced apoptosis. 31 In this study, we observed that Es-Cu did not significantly enhance autophagy under hypoxic conditions. However, under normoxic conditions, Es-Cu promoted cell death and suppressed malignant behaviors. Furthermore, the sensitivity of cells to Es-Cu-mediated cuproptosis, FDX1-driven, decreased, indicating resistance to cuproptosis. Nevertheless, the molecular mechanisms of linking hypoxia, copper ions, and autophagy in colorectal cancer require further investigation.
This study is the first to reveal the mechanism by which FDX1 regulates cuproptosis under hypoxic conditions to influence the malignant behavior of colorectal cancer. However, the potential synergistic interactions between FDX1 and other signaling pathways require further investigation. For instance, HIF-1α, a key regulator under hypoxia, may be closely associated with cuproptosis. A recent study by Wu et al. has demonstrated that improving the hypoxic tumor microenvironment enhances FDX1 expression and activates cuproptosis, thereby suppressing the initiation and metastasis of osteosarcoma. 32 This finding aligns with our results, further supporting the critical link between hypoxia and cuproptosis. Additionally, Joshi et al. have reported that lipoylation, a process occurring during cuproptosis, was not reversed under hypoxia, despite observed changes in cellular biological behavior. 33 Combined with our findings, we hypothesize that hypoxia may modulate tumor biological functions induced by cuproptosis through enhanced autophagy. Notably, FDX1 has been shown to inhibit the PI3K/AKT pathway, activating mitophagy and playing a pivotal role in hepatocellular carcinoma progression, 34 suggesting that FDX1 may regulate tumor cell fate via multiple signaling pathways. Therefore, future studies should explore the interactions between FDX1 and pathways such as HIF-1α, autophagy, and PI3K/AKT to unravel more complex regulatory networks. Collectively, targeting FDX1 and autophagy-related pathways may offer novel therapeutic strategies for colorectal cancer.
Conclusions
In summary, this study demonstrated that hypoxia induces autophagy, which suppresses FDX1-mediated cuproptosis, resulting in resistance to copper-induced cell death in colorectal cancer cells. The elucidation of this mechanism provided new insights and potential therapeutic targets for the treatment of colorectal cancer. However, the specific molecular pathways and signaling cascades underlying this process remain to be fully elucidated.
Limitation
This study has several limitations. 1) The sample size (N = 6 per group) was not determined by power analysis, which may affect the statistical reliability of the results. Future studies should expand cohort sizes and integrate power calculations to enhance robustness. 2) The precise molecular mechanisms linking FDX1 and autophagy remain incompletely elucidated. Further exploration of related signaling pathways (e.g., HIF-1α, mTOR, or AMPK) is needed to clarify their roles in hypoxia-induced cuproptosis tolerance. Addressing these limitations will deepen our understanding of FDX1-mediated regulatory networks and refine therapeutic strategies targeting cuproptosis in colorectal cancer.
Footnotes
Statements and declarations
Authors’ contributions
J.L. and L.Q. conceived and designed the study. Z.L., B.L. and M.L. collected samples and patient information. L.Q., Z.L. and B.Y. performed experiments, Y.T. and J.Y. analyzed the data, L.Q. wrote the manuscript. All authors have read and approved the final manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by Nanchong Science and Technology Bureau Major Chronic Disease Special Project (No. 23JCYJPT0018), and 2024 Provincial Financial Health Subsidy (clinical key specialty) (No. zk-2428-1).
Conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Patient consent for publication
Patients signed informed consent regarding publishing their data.
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper.