
Abstract
Background
Rhynchophylline (RIN) is an alkaloid known for its ability to effectively block signal transduction related to various neurodegenerative diseases. However, the specific mechanism by which RIN regulates microglial activation and cerebral ischemia remains unexplored. This study aims to investigate the function and molecular pathways through which RIN activates the JAK2/STAT3 signaling cascade, promoting the transformation of microglial phenotypes that contribute to recovery from cerebral ischemic injury.
Methods
By establishing a microglia oxygen glucose deprivation/reoxygenation (OGD/R) model and a middle cerebral artery occlusion animal model, we assessed changes in the expression of phenotype-specific marker factors for M1 and M2 microglia, as well as key proteins in the JAK2/STAT3 pathway, utilizing ELISA and Western blot techniques. Histological examination, including HE staining, TUNEL assay, and immunofluorescence, was employed to evaluate pathological changes in brain tissue, along with cell apoptosis and proliferation.
Results
The results indicated that microglial activity was significantly reduced and shifted towards the M1 phenotype following OGD/R. However, RIN treatment reversed these changes. When JAK2/STAT3 inhibitors were combined with RIN, it inhibited RIN’s protective effect. Animal studies have shown that RIN reduces histopathological changes associated with cerebral ischemia. Additionally, RIN inhibited microglial proliferation in ischemic cortical tissue and increased the expression of M2-type marker proteins, as well as the levels of phosphorylated JAK2 and STAT3 in the ischemic tissue.
Conclusion
In conclusion, this study indicates that RIN may protect against cerebral ischemic injury by activating the JAK2/STAT3 pathway, which promotes the transition of microglia to the M2 phenotypic.
Introduction
Cerebral ischemia is a leading cause of mortality in stroke patients globally, with significant implications for public health. 1 This condition triggers neurological deficits, which can progress to a more severe secondary complication known as cerebral ischemia-reperfusion injury (CIRI). CIRI exacerbates both mortality and disability among stroke survivors.2,3 Current estimates by the World Health Organization (WHO) suggest that approximately 30% of ischemic stroke patients do not survive the event. For those who do survive, the likelihood of developing various functional impairments and disabilities is high, profoundly impacting their quality of life. The incidence of ischemic stroke is particularly pronounced in the middle-aged and elderly population, aged 45 to 70 years, a demographic that is rapidly expanding due to the aging population trend in China. 4 While recombinant tissue plasminogen activator has emerged as the first of care for ischemic stroke treatment, its utility is constrained by a narrow therapeutic window, potential bleeding risks, and variable efficacy. 5 Given these limitations, there is an urgent and unmet need for the discovery and development of new pharmacological agents that can effectively promote the repair of cerebral ischemic injury. Such advancements are critical for enhancing the clinical management of ischemic stroke and improving outcomes for patients.
An accumulating body of research evidence suggests that inflammation and immune responses are pivotal in the pathogenesis of stroke, with studies indicating that the attenuation of these responses may improve outcomes following a cerebrovascular event. Prior research has underscored the importance of the microglial response in the inflammatory cascade that ensues post-stroke. 6 Microglia, the primary immune cells within the central nervous system (CNS), constitute approximately 5%–15% of the total cellular population in the adult brain 7. Under quiescent conditions, these resident microglia maintain a static state and are diffusely distributed throughout the CNS.8,9 However, the onset of cerebral ischemia triggers a rapid activation of microglia, characterized by marked morphological and functional alterations, culminating in the differentiation into two contrasting phenotypes: the pro-inflammatory M1 and the anti-inflammatory M2. 10 M1 microglia exacerbate inflammation through the secretion of cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor (TNF), while M2 microglia facilitate tissue repair by releasing anti-inflammatory mediators, including arginase-1 (Arg-1) and interleukin-10 (IL-10). 11 Following the initial phase of ischemia, microglia predominantly exhibit an M2 phenotype. However, with the persistence of ischemia, the release of inflammatory mediators prompts a shift towards the M1 phenotype, thereby intensifying the inflammatory response and exacerbating brain injury.12,13 Consequently, modulating the M1/M2 balance to direct the immune response towards a reparative state by promoting the conversion of microglia from the M1 to the M1 to the M2 phenotype is gaining recognition as a promising pharmacological intervention for cerebral ischemic injury. The Janus kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3) signaling pathway is a crucial pathway in the inflammatory process, playing a key role in the pathogenesis of neurological disorders, including cerebral ischemia and Parkinson’s disease. 14 Research has shown that STAT3 deficiency aggravates cerebral ischemic damage in both in vivo and in vitro models and also biases microglial polarization towards the M1 phenotype. 15
Rhynchophylline (RIN), a primary tetracycline alkaloid derived from constituent of Uncaria species, is widely used in the treatment of CNS disorders, including hypertension, convulsions, and strokes. RIN potently inhibits a diverse array of signal transduction pathways that are implicated in various degenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, epilepsy, and amyotrophic lateral sclerosis (ALS). 16 However, the literature provides only sparse information regarding the function of RIN in modulating microglial activation and neuroinflammation. Current research predominantly centers on the microglial inflammatory responses elicited by pharmacological agents such as lipopolysaccharide (LPS) or 2,5-dimethoxy-4-iodoamphetamine (DOI). Significantly, the capacity of RIN to regulate the microglial inflammatory cascade in the context of OGD/R has not been previously documented. To investigate this, the present study developed a hypoxia-glucose deprivation model using OGD/R in BV2 microglial cells and subsequently treated these cells with RIN to examine its potential influence on attenuating the microglial inflammatory response post-OGD/R. Additionally, a middle cerebral artery occlusion (MCAO) model was established to investigate the impact of RIN on the JAK2-STAT3 signaling pathway in microglial phenotypic transformation and its significance in the context of cerebral ischemia. The findings revealed that RIN promoted the transition of microglia towards the M2 phenotype, consequently diminishing the inflammatory response in the OGD/R model. Furthermore, both in vivo and in vitro studies disclosed that RIN elicited a shift in microglial phenotype towards the M2 subtype and reduced apoptosis through the activation of the JAK2-STAT3 pathway, thereby enhancing the repair process following brain injury.
Methods
Chemicals
RIN (purity: 99.94%), dexamethasone (purity: 99.86%), AG490 (purity: 99.97%), LPS, IFN-γ and IL-4 were purchased from Med. Chem Express (New Jersey, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), sugar-free DMEM, Fetal bovine serum (FBS) and trypsin were purchased from Thermo (Waltham, MA, USA). TNF-α, IL-1β, IL-6, IL-23, IL-10, TGF-β1, IL-13 and BDNF Enzyme-linked immunosorbent assay (ELISA) kits were purchased from Elabscience (Wuhan, China). Cell counting kit-8 (CCK-8 kit), BCA protein concentration assay Kit and One Step TUNEL Apoptosis Detection Kit were purchased from Beyotime (Shanghai, China). Total Protein Extraction Kit was purchased from Solarbio (Beijing, China). Rabbit monoclonal primary antibodies to Arg1, iNOS, p-JAK, p-STAT3, ki67 and iba1 were purchased from Abcam (Cambridge, UK). Mouse monoclonal primary antibody to CD163 was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Rabbit polyclonal primary antibody to CD86 was purchased from Affinity Biosciences (Jiangsu, China). HRP-conjugated monoclonal primary antibody to β-actin was purchased from Proteintech (Wuhan, China).
Cell culture
BV2 cells were obtained from Pricella in Wuhan, China. To induce the M1 subtype, the cells were treated with LPS and IFN-γ at a concentration of 10 ng/mL for 48 hours. For M2 subtype induction, IL-4 was administered at a concentration of 20 ng/mL for the same duration of 48 hours.
Microglia OGD/R model
BV2 cells were seeded into 96-well plates at a density of 8000 cells per well. After a cell monolayer developed, the medium in the wells was removed, and for the blank group, 100 μL of complete DMEM medium was added, while 100 μL of sugar-free DMEM medium was introduced to the model group. The cells were then incubated in a 5% CO2 cell culture incubator for 30 minutes before being transferred to a three-gas incubator, which provided a stable atmosphere of 95% N2 and 5% O2 at 37°C. They underwent oxygen and glucose deprivation for durations of 1, 2, 4, 6, and 8 hours. After the OGD period, the medium from each well was discarded and substituted with 100 μL of complete DMEM medium. Finally, the cells were returned to the 5% CO2 incubator for 24 hours to facilitate reoxygenation. Cell viability is assessed by determining the optical density (OD) at 450 nm for each cell group, with values expressed as a ratio of the OGD/R treated group to the control group (The control group is set up with cells that are normally cultured in a complete DMEM medium under conventional conditions, wherein the survival rate of the control group is regarded as 100%).
ELISA assay
Collected the supernatant from each group of cells and centrifuged at 4°C for 20 minutes at 1000×g. Collected the supernatant for subsequent testing. Added 100 µL of the samples to be tested into the appropriate wells of a 96-well ELISA plate, ensuring the setup of two duplicate wells for each sample. The plate was covered with a sealer and incubated for 90 minutes at 37°C. After the incubation period, each well was blotted dry, and 100 µL of biotinylated detection antibody working solution was added. Incubated at 37°C for 1 h. The plates were washed three times. Then, 100 µL of HRP enzyme conjugate working solution was added to each well. Incubated the plate at 37°C for 30 minutes, then washed each well three times. added 90 µL of the substrate solution (TMB) to each well and covered the plate. Incubated at 37°C for an additional 15 minutes. Added 50 µL of the termination solution to each well. Finally, measure the OD of each well at 450 nm using an enzyme marker immediately after adding the termination solution.
CCK-8 assay
BV2 cells were seeded into 96-well plates at a density of 1 × 10^4 cells per well, while various concentrations of RIN along with control groups were simultaneously established, comprising five wells for each condition. Once the cells adhered to the plate surfaces, different doses of RIN (2.0, 4.0, 8.0, 10, 20, 40, 100 μmol/L) were introduced to the RIN group. The cells were then incubated for 24 hours within a temperature and oxygen-controlled incubator. Subsequently, the OD was recorded at 450 nm using an Enzyme-labeled instrument. Cell survival rate was calculated using the formula: (absorbance of RIN-treat wells - absorbance of blank control wells) / (absorbance of control wells - absorbance of blank control wells) × 100%.
Western blot detection
Total proteins were extracted using a protein extraction kit, and the concentration of the proteins was measured with a BCA kit, followed by loading 50 μg of samples per well. SDS-PAGE gels with a 4%–20% gradient run for 1 hour before being transferred onto PVDF membranes, which were subsequently blocked with PBS buffer containing 5% skimmed milk powder for 1 hour. The primary antibody was incubated overnight at 4°C, while the secondary antibody was allowed to incubate at room temperature for 1 hour. For color development, ECL was utilized, and β-actin served as the internal reference. The target proteins were quantified for grey values using Image J.
Animal
7-week-old weighing 260-280 g male SD rats were obtained from Life River Laboratory Animal Technology Co. The Animal Ethics Committee of Kunming Medical University approved the study. A total of 40 rats were incorporated and the rats were randomly assigned to four groups: a sham operation group (n = 10), a cerebral ischemia group (n = 10), an anti-inflammatory agents treatment group (serving as a positive control) (n = 10), and a RIN treatment group (n = 10). Within the RIN treatment group, rats in the cerebral ischemia group received daily intraperitoneal injections of RIN (30 mg/kg) for seven consecutive days. Similarly, the anti-inflammatory treatment group was administered dexamethasone (5 mg/kg) using the same injection method every day for a week.
Establishment of rat CIRI model
Prior to surgical intervention, rats were housed in an environment maintained 22°C–25°C and were subjected to a 12-hour fasting period with ad libitum access to water. Animals were randomly assigned to four groups, each containing six individuals. Following the methodologies described by Koizumi 17 and Nagasawa, 18 a MCAO model with a 2-hour reperfusion interval was induced in the rat subjects. After weighing and anesthetizing the rats, they were secured on an anatomical frame, and the surgical site on the anterior neck was prepared by clipped the hair and disinfecting the skin. A 25 mm longitudinal incision was performed on the neck to expose and carefully dissect the right common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA). The ECA and CCA were then ligated, and an incision was made distal to the bifurcation of the ECA and ICA. A rounded-tip nylon suture, which had been heat-sterilized with an alcohol lamp, was inserted into the ICA to a depth of 20 ± 0.5 mm until resistance was encountered. Approximately 1 cm of the suture was left exterior to the vessel ligation site, the wound was treated with gentamicin, and then sutured closed. After 2 hours, the suture was carefully withdrawn to CCA level, ensuring that reperfusion of the compromised ICA and MCA was maintained through the CCA system, the basilar artery via the circle of Willis, and the contralateral external carotid artery. This maneuver completed the reperfusion phase of the MCA and the experimental protocol. In the sham operation group, only the CCA and ECA were ligated, and no suture was inserted. Throughout the 2 - hour ischemic period and the subsequent 1 - hour reperfusion, the rats' body temperature was maintained within the range of 36.5°C–37.5°C using a 40 - watt incandescent heat lamp. The sham operation group was identical to the experimental group except for the absence of suture insertion. Seven days following the surgical procedure, brain tissue and blood samples were procured from each group for analysis.
Zea-longa method
Following the ischemia and reperfusion period of 2 hours, rats were observed to be awake and exhibited neurological deficits. The neurological deficits subsequent to MCAO were assessed utilizing the Zea-longa scoring system 19 (0: no neurological impairment; one point: the front paws on the affected side cannot be completely extended; two points: the animal turns towards the affected side while ambulating; three points: the animal leans towards the affected side during walking; four points: cannot walk spontaneously and shows signs of loss of consciousness). They were tested once after modeling and again after administration. Animals that succumbed to anesthesia, surgical complications, or subarachnoid hemorrhage, as well as those that did not survive until the designated sampling time, were excluded from the experimental data analysis.
HE staining
Brain tissues were cut into tissue slices (thickness not exceeding 0.5 cm), put into a fixative solution for fixation and put into the embedding box, rinsed for 30 min, dewatered using different concentrations of alcohol, placed in xylene for transparency, and dipped in wax for embedding. The embedded sections were cut into thin slices using a slicer, placed on slides under hot water and dried. Xylene was utilized to extract paraffin from the sections, which were subsequently dehydrated with alcohol. The sections were immersed in an aqueous hematoxylin solution for 10 minutes, then stained with a mixture of acid water and ammonia for 5 seconds, followed by a 1-h rinse in running water. After dehydration with alcohol, the sections were stained with eosin solution for 2-3 minutes before undergoing dehydration and clarification. Lastly, drops of gum were applied, coverslips were affixed, and the samples were analyzed microscopically.
TUNEL staining
The TUNEL assay was conducted with the One-Step TUNEL Apoptosis Detection Kit (Green Fluorescence) supplied by Biyuntian Technology, based in Shanghai, China. The brain tissues from each group were gathered, and the paraffin sections underwent dewaxing and dehydration. The samples were treated with a protease K solution at a concentration of 20 μg/mL, maintaining a temperature of 37°C for 15 minutes, followed by washing twice with PBS for 5 minutes each. Next, a TUNEL reaction solution of 50 μL was applied dropwise and incubated at 37°C for 60 minutes, after which samples were washed three times with PBS for 5 minutes each. After this, 50 μL of converter - pod was introduced, and the samples were incubated at 37°C for 30 minutes, again followed by washing with PBS three times for 5 minutes each. Finally, the samples were sealed with an anti-fluorescence quencher and observed under a fluorescence microscope. Green fluorescence represents positive cells.
Immunohistochemical assay
The stripped tissue was sectioned, fixed, washed, dipped in wax, and embedded. Sections were placed in a Hydrogen Peroxide Block and incubated for 10-15 minutes. The buffer was washed twice for 5 minutes each. Ultra V Block was added dropwise and incubated for 5 minutes at room temperature to seal off non-specific background staining, followed by washing with buffer for 5 minutes twice. The primary antibody, anti-Ki67, was added dropwise and incubated at 37°C for 1-2 hours. The buffer was washed twice for 5 minutes each. HRP Polymer (enzyme secondary antibody) was added dropwise and incubated for 30 minutes at room temperature, followed by washing with buffer for 5 minutes twice. An appropriate amount of horseradish enzyme or alkaline phosphatase-labeled streptavidin working solution was then added dropwise and incubated at 37°C for 10 - 30 minutes. PBS was used to wash three times for 5 minutes each. The chromogen was developed for 3 - 15 minutes (DAB or NBT/BCIP). Finally, the sample was rinsed thoroughly, re-stained, sealed with film, and examined microscopically.
Immunofluorescence
Tissue sections were incubated at 4°C overnight with the primary antibody, while the cells were treated with goat anti-rabbit IgG secondary antibody for 1 hour. Subsequently, the nuclei were stained using DAPI. The percentage of Iba1-positive cells was measured with Image J software.
Statistical analysis
The statistical analysis utilized GraphPad Prism 9.0 software (San Diego, USA). Statistical significance was established at p < .05, while p < .01 indicated a highly significant result. The p-value was calculated using the independent t test and ANOVA. Each experimental data set comprised three replicates.
Results
Different OGD time and RIN concentration affected the survival of BV2 microglia
In comparision to the control group, exposure of BV2 cells to OGD for 1 h, 2 h, 4 h, 6 h, and 8 h resulted in a respective decline in cell viability of 15%, 28%, 46.1%, 58%, and 62%, demonstrating a clear time-dependent effect (Figure 1(a)). To validate the OGD/R model in microglia, subsequent experiments were conducted with an OGD duration of 4 h for BV2 cells. BV2 microglia were then treated with varying concentrations of RIN (2.0, 4.0, 8.0, 10, 20, 40, and 100 μmol/L) for 24 h, and the cell survival rates were determined using the CCK8 assay. The data revealed that RIN at concentrations of 2.0, 4.0, 8.0, 10, 20, or 40 μmol/L did not significantly affect the viability of BV2 microglia; however, a concentration of 100 μmol/L RIN markedly decreased the survival rate of microglia (p < .05) (Figure 1(b)). Therefore, a concentration of 40 μmol/L RIN was chosen for further investigation. Different OGD time and RIN concentration affect the survival of microglia (a), different OGD time affects the cell viability of BV2 microglia. (b), the survival rate of microglia in various concentrations of the RIN group. All data results are Mean ± SD, n = 3. Compared with the control group, *p < .05, **p < .01.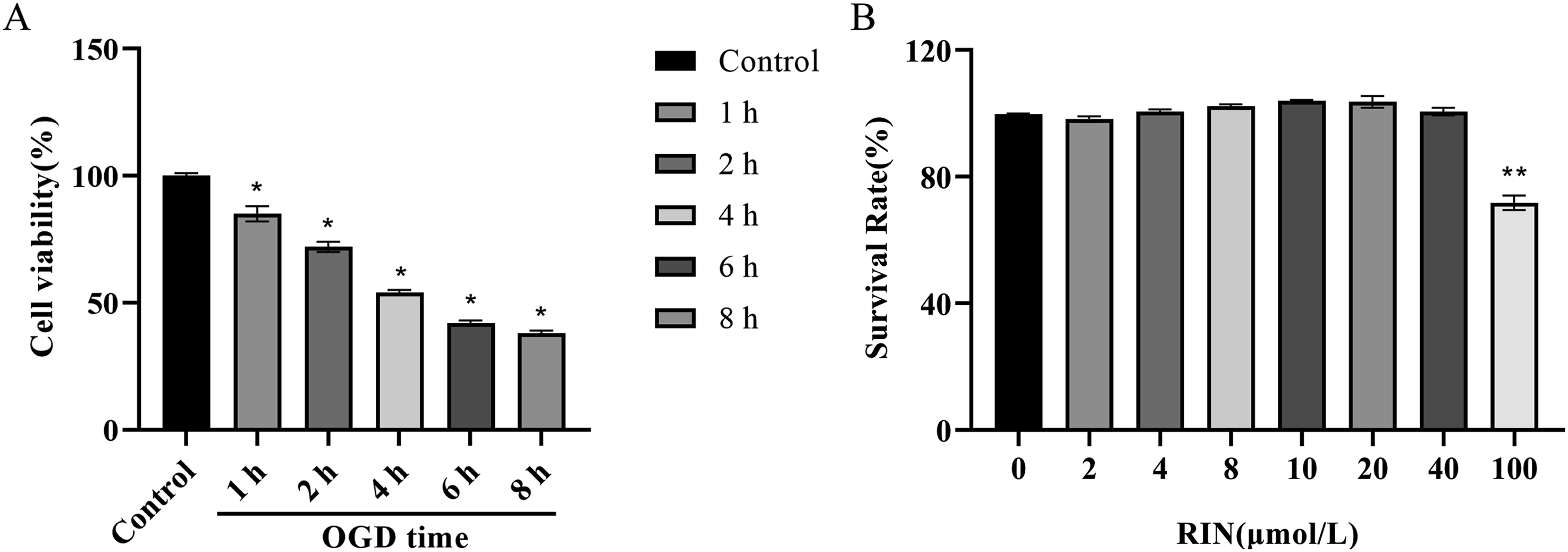
RIN induced phenotypic transformation of microglia M1 and M2 to regulate inflammatory response
To explore the association between RIN and the regulation of the inflammatory response in microglia, we treated RIN (40 μmol/L) to M1-type polarized microglia for a period of 24 hours. Western blot analysis indicated that the protein expression of CD86 and iNOS was significantly up-regulated in the M1 group compared to the control group. Conversely, the expression of Arg-1 and CD163 proteins was markedly enhanced in the M2 group, as well as in the RIN-treated and M1 + RIN-treated groups (Figure 2(a) and (b)). ELISA results illustrated that the secretion of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-23 was significantly higherin the M1 group than in the control group. In contrast, the secretion of these cytokines was reduced in the M2 group, the RIN treatment group, and the M1 + RIN group. Moreover, the M1 group showed a significant reduction in the secretion of anti-inflammatory cytokines, including IL-10, TGF-β1, and IL-13, compared to the control group, whereas the M2 group, the RIN treatment group, and M1 + RIN treatment group all demonstrated increased levels of these cytokines (Figure 2(c) and (d)). These data suggest that RIN may suppress the secretion of M1-associated inflammatory cytokines and promote the release of M2-type cytokines, thereby facilitating the transition of microglia to the M2 phenotype. The findings indicated that RIN induces a phenotypic shift in microglia from M1 to M2, thereby regulating the inflammatory response. The phenotype transformation of microglia M1 and M2 induced by RIN regulates inflammatory response (a) and (b), Western blot analyses were conducted to assess the levels of marker proteins associated with the differentiation of M1 and M2 microglia. (c) and (d), the cytokines M1 and M2 are detected by ELISA. All data results are Mean ± SD, n = 3. Statistically significant differences from the control group are indicated as #p < .05, ##p < .01, ###p < .001. Comparisons between the M1 and other groups are shown as *p < .05, **p < .01.
RIN could reduce inflammatory reaction via promoting M2 phenotypic transformation during OGD/R of microglia
In the subsequent experiment, normal BV2 cells were the negative control (NC) group. BV2 cells were exposed to an OGD/R protocol for 4 hours to establish the OGD/R model group. Additionally, OGD/R-treated cells were administered with RIN to constitute the treatment group. The CCK8 assay revealed a marked decline in microglial cell viability within the OGD/R model group compared to the NC group. However, RIN treatment significantly improved the viability of microglia post-OGD/R (Figure 3(a)). Western blot analysis showed that the expression of CD86 and iNOS was significantly elevated in the OGD/R model group compared to the NC group, whereas the expression of Arg-1 and CD163 was notably reduced. Notably, RIN treatment reversed the expression patterns of these proteins (Figures 3(b) and 3(c)). Furthermore, the OGD/R model group exhibited a substantial increase in the secretion of TNF-α, IL-1β, IL-6, and IL-23 relative to the NC group, accompanied by a significant decrease in the release of IL-10, TGF-β1, and IL-13; these alterations were mitigated following RIN treatment (Figure 3(d) and (e)). These findings further suggest that RIN may alleviate the inflammatory response by facilitating the transformation of microglia to the M2 phenotype during the OGD/R injury process. Rhynchophylline reduces inflammatory response by promoting M2 phenotypic transformation during OGD/R of microglia (a), The viability of microglial cells in various groups was assessed using the CCK8 assay. (b) and (c), The expression levels of marker proteins related to M1 and M2 microglial differentiation were analyzed via Western blot. (d) and (e), ELISA was employed to evaluate the levels of cytokines associated with M1 and M2 types. All data results are Mean ± SD, n = 3. In comparison to the NC group, #p < .05, ##p < .01, ###p < .001. When contrasted with the OGD/R group, *p < .05, **p < .01, ***p < .001.
RIN induced microglia to transform into M2 type by activating JAK2-STAT3 pathway
To gain further insight into the molecular mechanism by which RIN mediates the phenotypic shift in microglia, we designed a co-treatment experiment where M1-polarized microglia were exposed to a JAK2/STAT3 pathway inhibitor (AG490, 50 mmol/L) for 16 hours in conjunction with RIN treatment. Western blot analysis revealed that the M1 group exhibited a significant upregulation of CD86 and iNOS protein levels compared to the NC group, accompanied by a marked decrease in the expressions of phosphorylated p-JAK2 and p-STAT3 protein. In contrast, the M1 + RIN group showed a notable increase in the expressions of CD163, Arg-1, p-JAK2, and p-STAT3 proteins. Following RIN treatment, the subsequent addition of the JAK2-STAT3 pathway inhibitor led to a significant upsurge in CD86 and iNOS protein expression, while the expression of Arg-1, CD163, p-JAK2, and p-STAT3 proteins significantly declined (Figure 4(b) and (d)). Compared to the NC group, the M1 group showed significantly higher levels of TNF-α, IL-1β, IL-6, and IL-23 secretion, with a concurrent reduction in the secretion of IL-10, TGF-β1, and IL-13. RIN treatment reversed these patterns. However, when the JAK2-STAT3 pathway inhibitor was administered concurrently with RIN, the levels of these cytokines trended towards those observed in the M1 group (Figure 4(a) and (c)). These findings suggest that RIN may induce microglial transformation into the M2 phenotype by activating the JAK2-STAT3 pathway. RIN induces microglia to transform into M2 type by activating the JAK2-STAT3 pathway (a) and (c), Detection of M1 and M2 cytokines utilizing ELISA techniques. (b) and (d), Western blot analysis to assess the expression levels of marker proteins associated with M1 and M2 microglial differentiation. All data results are Mean ± SD, n = 3. Compared to the NC group, statistical significance was observed at #p < .05, ##p < .01, ###p < .001. In comparisons with the M1 group, significance levels were noted at *p < .05, **p < .01, ***p < .001, ****p < .0001. In comparison to the M1 + RIN group, + p < .05, ++ p < .01 indicates statistical significance.
RIN therapy diminishes microglia proliferation, alleviates neuronal apoptosis, and promotes brain injury repair
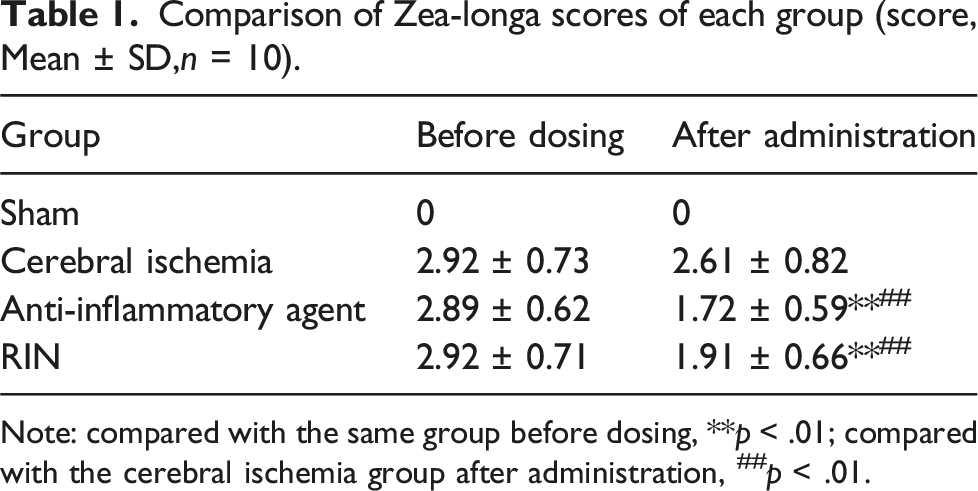
To substantiate the therapeutic potential of RIN in the context of cerebral ischemic injury, we developed a rat model of CIRI and carried out in vivo experiments to evaluate the efficacy of RIN. HE staining revealed that neurons in the sham-operated group were densely and orderly packed, displaying normal morphology and structure without any significant pathological alterations. In contrast, the ischemia group presented with neurons that were disarrayed and loosely packed, appearing contracted, with disrupted cell membranes, shrunken and hyperchromatic, and a marked reduction in the population of normal neurons. Compared to the cerebral ischemia group, both the anti-inflammatory drug-treated and RIN - treated groups exhibited a reduction in the extent of brain tissue damage, with neuronal morphology and quantity returning to near-normal levels (Figure 5(a)). There was no significant change in Zea-Longa scores before and following drug administration within the cerebral ischemia group; in contrast, a significant decline in scores was observed in both the anti-inflammatory drug-treated and the RIN - treated groups (p < .01). Furthermore, post-drug administration, the scores for both the anti-inflammatory drug group and the RIN treatment group were significantly lower than those of the cerebral ischemia group (p < .01) (Table 1, Figure 5(b)). TUNEL staining revealed a negligible number of apoptotic cells in the sham-operated group, whereas a substantial number of apoptotic cells were detected in the cerebral ischemia group. Importantly, the quantity of apoptotic cells showed a significant reduction in the rats treated with anti-inflammatory agents and RIN in comparison to the ischemic group (Figure 5(c)). RIN treatment diminishes microglia proliferation, alleviates neuronal apoptosis, and promotes brain injury repair (a), Histopathological changes were observed by HE staining. (b) Zea-Longa method was used to evaluate the neurological functions in rats. (c), TUNEL assay to assess nerve cell apoptosis within brain tissue. Comparison of Zea-longa scores of each group (score, Mean ± SD,n = 10). Note: compared with the same group before dosing, **p < .01; compared with the cerebral ischemia group after administration, ##p < .01.
Additionally, we investigated the impact of RIN treatment on the proliferation of microglia within the in vivo setting. As illustrated in Figure 5(d) and (e), we evaluated the expression of Ki67 and Iba1 in the cortical region of a CIRI rat model using immunohistochemistry and immunofluorescence assays. Compared to the sham-operated group, the ischemic group showed a marked upregulation of Ki67 and Iba1, indicative of a substantial increase in microglial proliferation after cerebral ischemia. Moreover, RIN treatment was observed to diminish microglial proliferation in the cortical regions. These results suggest that the enhanced proliferation of microglia in the cortical region, in conjunction with the increased neuronal apoptosis and aggravated brain tissue damage following cerebral ischemia, can be effectively attenuated by RIN treatment. Consequently, RIN contributes to the amelioration of brain tissue by concurrently reducing microglial proliferation and neuronal apoptosis.
RIN promotes microglia conversion to M2 phenotype by activating JAK2/STAT3 pathway to inhibit inflammatory response
Next, we examined the inflammatory response, phenotypic conversion, and the expression of crucial proteins in the JAK2/STAT3 pathway in rat brain tissue before and after RIN treatment. Tissue immunofluorescence staining results indicated that compared to the sham operation group, the cerebral ischemia group exhibited a significant increase in pro-inflammatory M1-type marker CD86-positive cells and a marked decrease in anti-inflammatory M2-type marker CD163-positive cells. Following treatment with anti-inflammatory drugs or RIN, there was a significant reduction in CD86-positive cells and a notable increase in CD163-positive cells when compared to the cerebral ischemia group (Figure 6(a)–(d)). Furthermore, Western blot experiments confirmed the impact of RIN treatment on the functional regulation of microglia in the brain tissue of rats with cerebral ischemia. Western blot analysis revealed that the expression levels of CD86 and iNOS were markedly elevated in the ischemic group compared to the sham group, whereas the expressions of Arg-1, CD163, p-JAK2, and p-STAT3 was significantly decreased. Following treatment with RIN, a reversal in the expression of these proteins was observed (Figure 6(e) and (f)), which was similar to the results obtained with the positive control (anti-inflammatory agents). Additionally, the cerebral ischemia group, unlike the sham group, exhibited increased levels of the pro-inflammatory cytokines IL-1β and IL-6, as well as enhanced levels of the anti-inflammatory cytokines IL-10 and BDNF in brain homogenates. In contrast, the pro-inflammatory cytokines IL-1β and IL-6 were downregulated, and the anti-inflammatory cytokines IL-10 and BDNF were upregulated in both the anti-inflammatory agents group and the RIN-treated group compared to the ischemic group (Figure 6(g)). These findings suggest that RIN treatment may facilitate the transition of microglia to the M2 phenotype, thereby reducing inflammation and immune response, and promoting the repair process of brain injury. RIN encourages the transformation of microglia into the M2 phenotype by activating the JAK2-STAT3 signaling pathway, which suppresses the inflammatory response (a)-(d), Representative immunofluorescence images of CD86-positive and CD163-positive cells in the cerebral cortex tissue of rats. (e) and (f), Expression of M1 and M2 microglia phenotypic markers and key proteins of the JAK2/STAT3 signaling pathway in rat brain tissue were determined by Western blot analysis. (g), The contents of inflammatory factors IL-1β, IL-6, IL-10, and BDNF in brain homogenate of rats from each experimental group, performed using ELISA. All data results are Mean ± SD, n = 3. When compared to the control group, ##p < .01, ###p < .001. In contrast to the Cerebral ischemia group, *p < .05, **p < .01, ***p < .001.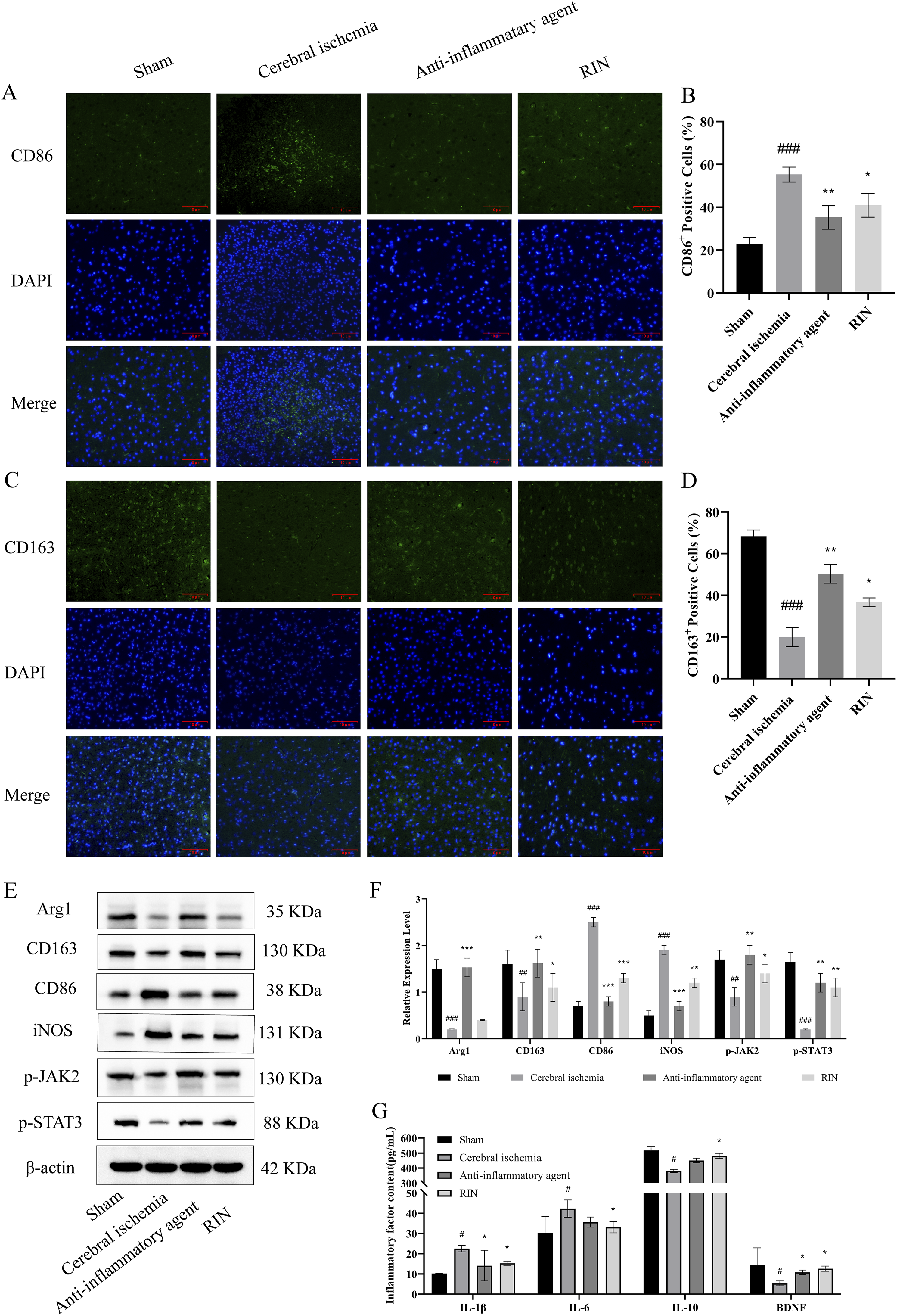
Discussion
The pathogenesis of CIRI is profoundly complex, and significant progress in clinical interventions has been elusive. In response to physical trauma or various in vivo stimuli, microglial cells become hyperactive, adopting a polarized phenotype that is heavily influenced by the characteristics and intensity of the insult, thereby significantly affecting the course of the disease. Nonetheless, the molecular cascades triggering microglial activation are not fully elucidated. Additionally, the polarized state of microglia confers both beneficial and detrimental effects. Gaining a deeper comprehension of how microglia are activated and finding a way to balance the M1 and M2 phenotypes could provide novel therapeutic avenues for the clinical handling of CIRI. Such insights may be crucial for realizing transformative advancements in treatment strategies.
Studies have demonstrated that RIN confers a protective effect on both neuronal and glial cells. Furthermore, RIN has been shown to diminish brain injury in the permanent middle cerebral artery occlusion (pMCAO) stroke model. 20 RIN markedly enhances the behavioral alterations in tail suspension (TS) rats induced by the hallucinogen DOI, reduces the concentrations of inflammatory cytokines in serum and striatum, and suppresses the inflammatory response in BV2 microglia challenged with DOI by inhibiting the TLR/NLRP3/NF-κB signaling pathway. 21 In addition, RIN attenuates LPS - induced inflammation in primary microglia cells by down-regulating the MAPK/NF-κB signaling pathway. 22
Microglia serve as the principal immune sentinels within the CNS, adept at recognizing danger-associated molecular patterns and rapidly responding to protect the brain from injury by adopting diverse functional phenotypes, notably the M1 and M2 polarized types. 23 These two polarized phenotypes of microglia represent a spectrum of dynamic changes in their activation status. M1 polarization leads to the release of pro-inflammatory mediators, which can impede CNS recovery, whereas the M2 polarization is associated with the secretion of anti-inflammatory cytokines, thereby reducing CNS damage and facilitating repair. These highly plastic cells are capable of swiftly transitioning between activation states. In pathological conditions, activated microglia maintain a dynamic balance between M1 and M2 polarization, rendering the M1/M2 ratio a pivotal factor in predicting stroke outcomes in the context of brain injury.24,25 Kanazawa et al. reported the detection of M2 microglia 12 hours after cerebral ischemia, with their numbers increasing to a maximum at 3 days post-ischemia before declining. Conversely, M1 microglia were observed to increase within the first 14 days following the ischemic insult. 26 Consequently, the identification of pharmacological agents capable of inhibiting M1 polarization or promoting a shift towards M2 could alleviate oxidative stress and inflammatory subsequent to cerebral ischemia, ultimately enhancing neurological recovery. In the present study, we observed that following the establishment of the OGD model, microglial morphology transitioned from a ramified to a rounded phenotype, indicative of activation in response to the OGD condition. Additionally, ELISA results demonstrated an increase in inflammatory factor secretion during this phase, suggesting a shift from a quiescent to an M1 phenotype. Subsequent administration of RIN induced a conversion towards the M2 phenotype.
Shadamu et al. 27 employed a rat model to simulate in vivo using MCAO and utilized BV2 cells for in vitro experimentation via the OGD model. Their research indicated a marked up-regulation of both M1 and M2 markers, reinforcing the concept that manipulating microglial polarization could mitigate inflammatory responses and protect neuronal cells. In our study, we evaluated the expression levels of CD86, iNOS, CD163, and Arg-1 in ischemic brain tissues using Western blot analysis. Additionally, we employed ELISA to quantify the concentrations of the inflammatory cytokines IL-1β, IL-6, and the anti-inflammatory cytokine IL-10, as well as BDNF, a crucial factor of tissue repair, in rat brain homogenates. The data revealed that RIN potently down-regulated the expression of CD86 and iNOS, inhibited the secretion of pro-inflammatory cytokines IL-1β and IL-6, and concurrently up-regulated the levels of CD163 and Arg-1. Moreover, RIN significantly elevated the synthesis of IL-10 and BDNF, thereby promoting a shift in microglial polarization from the M1 to the M2 phenotype. Our findings confirmed that RIN enhances the viability of OGD/R-treated microglia and reduces cellular injury. Additionally, animal studies demonstrated that this compound alleviate cerebral ischemia/reperfusion damage in rats following MCAO. The proposed mechanism of action involves the modulation of microglial M1/M2 polarization, suppression of the central inflammatory response, and augmentation of restorative processes within the post-ischemic brain tissue.
In addition, we investigated the role of RIN in facilitating the transition of microglia to the M2 phenotype and its contribution to the recuperation from cerebral ischemic injuries via the JAK2/STAT3 signaling pathway. This pathway is typically quiescent under normal physiological conditions but is indispensable for CNS development. 28 This pathway is activated in response to ischemic stress and interacts with various pathogenic elements, including apoptosis and inflammation. 29 Numerous prior investigations have indicated that the regulation of the JAK2/STAT3 signaling pathway is regulated bidirectionally. Studies indicate that the activation of this pathway promotes the differentiation of microglia into the anti-inflammatory M2 phenotype, thereby contributing to the prevention of further neurological damage.30,31 Conversely, other studies suggests that blocking STAT3 activation can alleviate neuronal injury.32,33 In our research, we assessed the expression levels of key proteins within the JAK2/STAT3 pathway using Western blot analysis in both in vitro and in vivo models. Our findings reveal that RIN treatment is associated with elevated levels of M2-type anti-inflammatory factors, coupled with a significant increase in the expression of p-JAK2 and p-STAT3 when compared to the OGD/R or CIRI model groups. Moreover, the co-administration of RIN with a JAK2/STAT3 inhibitor in the OGD/R model resulted in a reduction in the expression of p-JAK2 and p-STAT3. To summarize, this study illustrates that RIN can stimulate phenotypic shifts in neuromicroglia and promote recovery following cerebral ischemic injury through the JAK2/STAT3 signaling pathway. However, the observed heterogeneity in the JAK2/STAT3 pathway responses in previous stroke research may be influenced by a variety of factors. Consequently, further studies are necessary to elucidate the precise functions of JAK2 and STAT3 in the pathogenesis of stroke, which will be instrumental in developing novel therapeutic strategies for ischemic stroke.
Conclusion
In this research, we examined the role of RIN in facilitating the phenotypic transformation of neuromicroglia by engaging the JAK2/STAT3 signaling pathway, utilizing both an in vitro cellular model and an in vivo animal model. This engagement serves to hinder the detrimental inflammatory response within brain tissues and improves brain injury that occurs after cerebral ischemia.
Footnotes
Acknowledgments
We extend our gratitude to all the researchers who played a role in this study.
Author contributions
PB was instrumental in formulating the study, conceptualizing the methodology, analyzing or synthesizing the research data, and drafting the manuscript. CL and LY played vital roles in analyzing and preparing the manuscript. YL and MJ were responsible for carrying out the experiments, whereas LW contributed to the analysis by engaging in constructive discussions and reviewing and refining the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research received financial support from the Joint Special Project of the Yunnan Science and Technology Department and Kunming Medical University (202101AY070001-094), as well as from the Sub-project of the Audit System Disease Clinical Pharmacy Center in 2022 (ZX2019030501).