
Abstract
Introduction
Post-stroke depression (PSD) is one of the common complications after stroke. Trimethylamine-N oxide (TMAO) has been reported to exacerbate brain injury after ischemic stroke, and its expression is positively related to the severity of depressive symptoms. This study was performed to investigate the role and mechanism of TMAO in PSD.
Methods
The classical stroke model was combined with spatial constraint stress to establish a PSD mouse model, and the effect of TMAO on the depression-like behavior of the PSD mouse model was evaluated using the tail suspension test and the forced swimming test. ELISA was employed to measure the concentration of TMAO and cortisol. RT-qPCR, Western blot, and Immunohistochemistry assay were implemented to determine mRNA and protein expression. Endothelial permeability was assessed using the fluorescein isothiocyanate-dextran permeability assay in vitro. Reactive oxygen species (ROS) level was examined with a ROS detection kit.
Results
TMAO dose-dependently exacerbated depression-like behavior, induced the activation of the p38/mitogen-activated protein kinase (MAPK) signaling pathway, downregulated the abundance of tight junction proteins, and resulted in the dysregulation of neurotrophic mediators in the PSD mouse model in vivo. Further, TMAO increased endothelial permeability and activated reactive oxygen species-p38/MAPK signaling. By introducing the p38/MAPK pathway inhibitor SB203580, TMAO was found to downregulate the expressions of tight junction proteins and promoted endothelial permeability via p38/MAPK signaling activation.
Discussion
By activating the ROS-P38/MAPK pathway, TMAO enhanced the permeability of the blood–brain barrier after stroke. These mechanisms resulted in the dysregulation of neurotrophic mediators, ultimately leading to PSD progression.
Introduction
Stroke is a cerebrovascular disease associated with high morbidity and mortality rates globally. Approximately one-third of patients who survive experience depression within the first 5 years after stroke. 1 The main clinical manifestations include retardation of thinking and lack of interest. Due to its adverse impact on the prognosis of patients with stroke, post-stroke depression (PSD) is a significant public health issue. Previous studies reported that the main pathophysiological mechanisms of PSD are changes in depressive neural circuits caused by white matter atrophy.2,3 Therefore, to prevent and treat PSD, it is prominently important to understand its pathogenesis.
The microbiome–gut–brain axis refers to bidirectional signaling between the gut microbiota and central nervous system. 4 The gut microbiota, also known as the second brain, may influence brain homeostasis via the microbiome–gut–brain axis under physiological and pathological conditions. Accumulating evidence has reported that the gut microbiota affects brain function and behavior, which can lead to conditions including anxiety, depression, and decreased cognition.5–8 For example, Shao et al. reported that gut microbiota might be implicated in PSD progression, with mechanisms associated with the MAPK signaling pathway. 9 In addition, Luo et al. reported that there was a prominent alteration of gut microbiota composition at the phylum, family, and genus levels between PSD patients and healthy individuals. 10
Trimethylamine-N-oxide (TMAO) is a small organic compound derived from the oxidation of trimethylamine, which is primarily produced by gut microbiota from dietary precursors such as choline, phosphatidylcholine, and L-carnitine. In recent years, the role of TMAO has been increasingly studied in neurological disorders. Previous studies reported that elevated level of TMAO has been associated with neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease through the biological processes of oxidative stress and inflammation.11,12 In stroke, TMAO has been reported to exacerbate brain injury after ischemic stroke by multiple pathological processes, including accelerating atherosclerosis, enhancing thrombogenesis potential, promoting vascular inflammation and endothelial dysfunction, and increasing oxidative stress. 13 In addition, TMAO might be involved in PSD through its effects on inflammation and the central nervous system. 14 The pro-inflammatory properties of TMAO could potentially worsen neurological outcomes by impacting neurotransmitter systems and neural plasticity, which were vital in mood regulation and recovery post-stroke. These studies emphasized the importance of TMAO in the pathophysiology of stroke and its potential as a target for therapeutic intervention to improve outcomes. However, the role of TMAO in PSD progression and its potential molecular mechanism are not fully elucidated.
In summary, previous study has suggested a possible correlation between TMAO and PSD. This study aimed to investigate the relationship between TMAO and PSD in human samples and PSD mouse model. In addition, brain microvascular endothelial cell line (hCMEC/D3) was utilized to explore the underlying molecular mechanisms.
Materials and methods
Animals
In total, 20 BALB/c mice were acquired from Jiangsu Aniphe Biolaboratory (Jiangsu, China) and were raised adaptively for 1 week under standard environment conditions at a temperature of 20°C–25°C and 12-h light/dark cycle. The procedure in the animal experiment was ratified by the Laboratory Animal Committee of Wuhan Wuchang Hospital. It was performed in accordance with the National Natural Science Foundation guidelines for the care and use of Laboratory animals.
Establishment of the PSD mouse model
Patients with hemiplegia after ischemic stroke are highly dependent on caregivers due to mobility difficulties, which can lead to the development of depressive symptoms. To simulate the clinical course of PSD, an animal model combining the classic stroke model (middle cerebral artery occlusion [MCAO]) and spatial constraint stress was established.
15
The mice were arbitrarily divided into the sham, PSD, PSD + low-dose TMAO, and PSD + high-dose TMAO groups, and each group included five mice. The MCAO stroke model was established using an established protocol.
16
The blood supply was restored by withdrawing the suture after MCAO for 60 min. The mice in the PSD group were subjected to spatial constraint stress for 2 h/day for 2 weeks from the 4th day after MCAO. Spatial constraint stress was achieved by placing the mice in a 50-mL centrifuge tube. A 50-mL centrifuge tube that had been modified for improved ventilation was used in the experiment. Thus, the mice could not move forward or backward. The mice in the sham group underwent the same surgical procedures for MCAO, except that the left middle cerebral artery was not occluded. Further, they were not subjected to spatial constraint stress. The mice in sham and PSD groups were fed with standard diet. The mice in the PSD + low-dose TMAO group were fed with standard diet containing 0.1% TMAO (1 g/kg; Sigma, St. Louis, MO, the USA; catalogue no. #317594; purity≥95%) for 8 weeks. The mice in the PSD + high-dose TMAO group were fed with standard diet containing 0.3% TMAO (3 g/kg; Sigma) for 8 weeks.
17
At the end of the experimental period, behavior assessments were performed. Then, the mice were euthanized with carbon dioxide, and the mouse blood samples were collected from the abdominal aorta. The blood samples were centrifuged at 4000 g for 10 min at 4°C to obtain serum. Serum samples were stored at −80°C and serum TMAO and cortisol concentrations were detected by enzyme-linked immunosorbent assay (ELISA). Brain tissues were collected and a part of brain tissues were stored at −80°C for the following real time quantitative polymerase chain reaction (RT-qPCR), Western blot, and ELISA analyses. Brain tissue blocks were paraffin-fixed for immunohistochemistry (IHC) assay. The complete time flow of the animal experiment is shown in Figure 1(a). High dose of TMAO prominently aggravates depression-like behavior in PSD mice model. BALB/c mice were divided into four groups: Sham, PSD, PSD + TMAO (low dose), and PSD + TMAO (high dose). (a) The experimental procedure and timeline for the mouse model was shown. The MCAO stroke model was established at Day 1, and the mice were subjected to spatial constraint stress for 2 h/day at Day 5-Day 18. The mice were euthanized after feeding diet containing different doses of TMAO at Day 19-Day 74, and the brain and blood samples of all mice were collected. (b and c) Tail suspension test (TST) and forced swimming test (FST) were conducted to assess depression-like behavior in mice. (d and e) The level of TMAO in the serum and brain tissues was analyzed by ELISA. (f) The serum cortisol level was measured by ELISA. *p < .05 vs. Sham group, ***p < .001 vs. Sham group, #p < .05 vs. PSD group, ##p < .01 vs. PSD group, ###p < .001 vs. PSD group.
Tail suspension test
The tail suspension test (TST) was conducted to evaluate the depression-like behavior of mice. 18 The TST was carried out on a bracket 35 cm away from the base. A hook was fixed on the bracket, and the mouse tail was fixed approximately 1 cm away from the origin. Before the test, the mice entered the room 1 h earlier to adapt to the environment. The test lasted for 6 min, of which the first 2 min were for acclimatization, and the total duration of immobility was measured during the following 4 min.
Forced swimming test
The forced swimming test (FST) was carried out based on the protocol outlined in a previous study. 19 Mice were placed in a glass cylindrical container measuring 20 cm in height and 15 cm in diameter for a duration of 6 min. After 2 min of acclimatization, the total duration of immobility was measured during the following 4 min of testing. The container was filled with water at a temperature ranging from 23°C to 25°C, with a depth of 14 cm. The duration of immobility was measured if the mice floated or made minimum movement necessary to maintain floating in the water. During the experiment, the mental state and physiological state of the mice should be keenly observed, and the experiment should be terminated immediately if necessary to prevent the mice from drowning.
Measurement of TMAO concentration
The concentration of TMAO in the mouse serum and brain tissue samples and human serum samples was determined using the enzyme-linked immunosorbent assay kit (ELK Biotechnology; catalogue no.:ELK8678), and the testing process strictly followed the kit instructions. The optical density at 450 nm was read via a microplate absorbance reader.
Measurement of serum cortisol concentration
The serum cortisol concentration was determined via the enzyme-linked immunosorbent assay kit (Mlbio, Shanghai, China; catalogue no.: ml001959), and the testing process strictly followed the kit instructions. The optical density at 450 nm was read via a microplate absorbance reader.
Human blood samples collection
The blood samples from 15 healthy controls, 15 stroke patients without depression, and 15 PSD patients were collected, and the serum was isolated. The procedure conform to the Declaration of Helsinki regarding human experimentation. The procedure is approved by the Medical Ethics Committee of Wuhan Wuchang Hospital.
Hamilton rating scale for depression (HAMD)
Depressive severity of 15 PSD patients was evaluated by the HAMD. 20
RT-qPCR
RT-qPCR was performed using the Trizol reagent (Beyotime, Shanghai, China) to isolate RNA samples from the mice brain tissues and hCMEC/D3 cells. Reverse transcription was conducted using the PrimeScript RT Reagent Kit (Takara, Dalian, China). Amplification reaction was conducted via the SYBR Green qRT-PCR Kit (Takara). GAPDH was used as the housekeeping gene, and the relative abundance was analyzed via the relative quantification (2−ΔΔCt). occludin (mouse) – forward primer: 5’-CCACCCCCATCTGACTATGC-3’, reverse primer: 5’-TCGCTTGCCATTCACTTTGC-3’; claudin-1 (mouse) – forward primer: 5’-GGCTTCTCTGGGATGGATCG-3’, reverse primer: 5’-CCCCAGCAGGATGCCAATTA-3’; claudin-5 (mouse) – forward primer: 5’-GTTAAGGCACGGGTAGCACT-3’, reverse primer: 5’-TACTTCTGTGACACCGGCAC-3’; ZO-1 (mouse) – forward primer: 5’-TGAACGTCCCTGACCTTTCG-3’, reverse primer: 5’-CTGTGGAGACTGCGTGGAAT-3’; ZO-3 (mouse) – forward primer: 5’-CGACTATGAGGACACCGACG-3’, reverse primer: 5’-TTGTCCCATGACCCATCAGC-3’; GAPDH (mouse) – forward primer: 5’-TGGAAAGCTGTGGCGTGAT-3’, reverse primer: 5’-GTTGCTGTTGAAGTCGCAGG-3’; occludin (human) – forward primer: 5’-AGCAGCGGTGGTAACTTTGA-3’, reverse primer: 5’-CCTCCAGCTCATCACAGGAC-3’; ZO-1 (human) – forward primer: 5’-TGCCTCTGAGAGAGACGACA-3’, reverse primer: 5’-GGAGGCCTATCGTGTGATCG-3’; GAPDH (human) – forward primer: 5’-GGAAGCTTGTCATCAATGGAAATC-3’, reverse primer: 5’-TGATGACCCTTTTGGCTCCC-3’.
Western blot assay
The brain tissue samples of mice and hCMEC/D3 cells were disrupted with the RIPA buffer (Beyotime, Shanghai, China). The protein sample concentrations were measured via the BCA Protein Assay Kit (Beyotime). Protein samples were loaded onto the SDS-PAGE and transferred onto the PVDF membrane (Millipore, Billerica, MA, the USA). After blocking the non-specific sites by incubating with 5% skimmed milk for 1 h, the membranes were mixed with the diluted primary antibodies overnight. The primary antibodies utilized were as follows: anti-occludin (Proteintech, Wuhan, China; catalogue no.: 27260-1-AP; 1:8000), anti-claudin-5 (Proteintech; catalogue no.: 29767-1-AP; 1:8000), anti-ZO-1 (Proteintech; catalogue no.: 21773-1-AP; 1:8000), anti-ZO-3 (Abcam, Boston, MA, the USA; catalogue no.: ab191143; 1:5000), anti-p38 (Proteintech; catalogue no.: 14064-1-AP; 1:5000), anti-p-p38 (Proteintech; catalogue no.: 28796-1-AP; 1:3000), anti-Bax (Proteintech; catalogue no.: 50599-2-Ig; 1:10000), and anti-Bcl-2 (Proteintech; catalogue no.: 26593-1-AP; 1:3000). On the next day, the membranes were labeled with the diluted HRP-conjugated secondary antibody for 2 h. The protein bands were visualized using the ECL kit (Beyotime), and protein quantification was carried out via ImageJ (NIH, Bethesda, MD, the USA).
Immunohistochemistry assay
The brain tissue samples of mice were subjected to immunohistochemistry assay to measure the levels of brain-derived neurotrophic factor (BDNF) and glial fibrillary acidic protein (GFAP). Briefly, the tissue samples were immobilized with 4% paraformaldehyde, paraffin-embedded with 100% liquid paraffin, and sliced into 5-µm-thick sections. The sections were mixed with phosphate-buffered saline solution/Tween plus 0.1% H2O2 for 30 min to block the endogenous peroxidase, followed by mixture with 1.5% normal goat serum for 30 min. The sections were then mixed with anti-BDNF (Santa Cruz Biotechnology, Santa Cruz, CA, the USA; 1:500) or anti-GFAP (Santa Cruz Biotechnology, Santa Cruz, CA, the USA; 1:300). Then, the sections were mixed with peroxidase conjugated secondary antibody. The abundance of BDNF and GFAP was analyzed using Image J (scale bar: 50 µm).
Cell culture and grouping
hCMEC/D3 cell line was maintained in the endothelial cell medium (ScienCell; catalogue no. #1001). TMAO was purchased from Sigma (catalogue no. #317594) and was dissolved in dimethylsulfoxide. In Figures 4 and 5, hCMEC/D3 cells were incubated with TMAO at doses of 0, 100, 300, 600, or 900 μM for 24 h. In Figure 6, hCMEC/D3 cells were divided into the control (dimethylsulfoxide of the corresponding volume was added to the medium), TMAO (900 μM; 24 h), and TMAO + SB203580 (2 μM, 24 h) groups.
Cell counting kit-8 assay
Cell viability was evaluated via the cell counting kit-8 assay. hCMEC/D3 cells were re-suspended at the concentration of 5 × 105 cells/mL, and 100 µL of cell suspension was added to each well of 96-well plates. The next day, cells were exposed to gradient concentrations of TMAO (0, 100, 200, 300, 600, 900, 1200, or 2000 μM) for 24 h or 900 μM TMAO for 0, 4, 8, 12, 24, or 48 h. The cell counting kit-8 reagent (Beyotime) was pipetted to the wells to incubate with hCMEC/D3 cells for 4 h at 37°C. The absorbances values were read at the wavelength of 450 nm.
Fluorescein isothiocyanate-dextran permeability assay
BBB disruption was evaluated using the endothelial cell monolayer fluorescein isothiocyanate (FITC)-dextran permeability assay. hCMEC/D3 cells were plated onto the 0.4-µm Pore Polycarbonate Membrane Inserts in 6.5-mm Transwell® plates (Corning, New York, NY, the USA) at 2 × 104 cells/well. Further, they were allowed to grow for confluence. After exposure to gradient concentrations of TMAO (0, 100, 300, 600, or 900 μM) for 24 h, endothelial permeability was evaluated via FITC-dextran. In particular, 20 µg/mL of FITC-dextran (Sigma-Aldrich) was introduced into the above compartments. Meanwhile, fresh PBS solution was pipetted to the below compartments. Subsequently, the system was incubated for 30 min, thereby allowing the measurement of the amount of FITC-dextran that passed through the damaged junctional integrity of the membrane insert into the lower compartment. Fluorescence (excitation: 490 nm and emission: 520 nm) was determined using a fluorimeter.
ROS activity analysis
The commercial ROS detection kit (Beyotime; catalogue no. S0033S) was used to analyze ROS activity. hCMEC/D3 cells were re-suspended at the concentration of 5 × 105 cells/mL, and 100 µL of cell suspension was added to each well of 96-well plates. The next day, cells were exposed to gradient concentrations of TMAO (0, 100, 300, 600, or 900 μM) for 24 h. Dichlorodihydrofluorescein diacetate was diluted at a ratio of 1:1000 and was administered at 10 μmol/L. The cell culture supernatant was subsequently removed, and the dichlorodihydrofluorescein diacetate reagent was introduced to incubate with hCMEC/D3 cells for 40 min at 37°C. Thereafter, the cells were rinsed thrice using the serum-free medium. The mean fluorescence intensity of ROS was determined.
Data analysis
Data were processed via the GraphPad Prism 8.0 software (GraphPad Prism, La Jolla, CA, the USA) and were indicated as mean ± standard deviation. All experiments were conducted in triplicates unless otherwise stated. One-way analysis of variance was utilized to analyze the differences in multiple groups. A p value of <0.05 indicated statistically significant differences.
Results
High-dose TMAO prominently aggravates depression-like behavior in the PSD mouse model
We first established PSD mouse model to explore the role of TMAO in vivo. The experiment workflow for the mouse study is shown in Figure 1(a). PSD mouse model was established using the classic MCAO stroke model followed by spatial constraint stress, and different doses of TMAO were administrated for 8 weeks before behavior assessment and sample collection. The PSD group had a longer immobility time based on the TST and FST than the sham group (Figures 1(b) and 1(c)), which was interpreted as depression-like behavior, suggesting the successful establishment of the PSD mouse model. TMAO intensified depressive-like behaviors in mice in a dose-dependent manner (Figures 1(b) and 1(c)). The serum and brain tissue TMAO levels of the PSD group were prominently upregulated compared with those of the sham group (Figures 1(d) and 1(e)). Moreover, the addition of TMAO dose-dependently elevated its level in both the serum and brain tissue samples (Figures 1(d) and 1(e)), suggesting the successful administration of TMAO. The serum cortisol abundance of the PSD group was prominently elevated compared with that of the sham group (Figure 1(f)), suggesting that the PSD mouse model presented with neuroendocrine disturbance. In addition, TMAO further increased the serum cortisol concentration in a dose-dependent way (Figure 1(f)). In conclusion, high-dose TMAO exacerbated depression-like behaviors in the PSD mouse model.
To further confirm the relationship between TMAO and PSD, we assessed the serum TMAO level in healthy controls, stroke patients, and PSD patients. TMAO abundance of the stroke group was prominently upregulated compared with the healthy control group. In addition, the TMAO abundance of the PSD group was further enhanced compared with the stroke group (Supplemental Figure 1(a)). There was a significant positive correlation between the TMAO level and the Hamilton Depression Rating Scale score in patients with PSD (Supplemental Figure 1(b)). These data further demonstrated that TMAO had a pivotal role in the development of PSD.
TMAO dose-dependently downregulates the abundance of tight junction proteins and activates the p38 signaling pathway in the PSD mouse model
The mRNA abundance of five tight junction proteins (occludin, claudin-1, claudin-5, ZO-1, and ZO-3) in the PSD group prominently reduced. This phenomenon was further aggravated by the administration of TMAO in a dose-dependent way (Figures. 2(a)–2(e)). Similarly, the protein levels of tight junction proteins also presented with a consistent trend (Figures. 2(f)–2(j)). To explore whether the p38 signaling modulated the TMAO-mediated disruption of tight junction proteins, the total p38 and phosphorylated p38 levels were measured. The p-p38/p38 level of the PSD group prominently increased compared with the sham group. This phenomenon was further exacerbated by TMAO in a dose-dependent manner (Figures 2(f) and 2(k)). Therefore, TMAO further activated the p38 signaling pathway in the PSD mouse model. Further, the PSD-induced upregulation of pro-apoptotic protein Bax and the downregulation of anti-apoptotic protein Bcl-2 were aggravated by TMAO (Figures 2(f), 2(l), and 2(m)). Taken together, TMAO activated the p38 signaling pathway and downregulated the abundance of tight junction proteins in the PSD mouse model. TMAO dose-dependently downregulates tight junction proteins and activates p38 signaling pathway. (a-e) RT-qPCR was performed to determine the mRNA expression of tight junction proteins in mouse brain tissues, including occludin, claudin-1, claudin-5, ZO-1, and ZO-3. (f-m) The levels of tight junction proteins (occludin, claudin-5, ZO-1, and ZO-3), p-p38, p38, and two apoptosis-associated proteins (Bax and Bcl-2) were examined by western blot assay. **p < .01 vs. Sham group, ***p < .001 vs. Sham group, #p < .05 vs. PSD group, ##p < .01 vs. PSD group, ###p < .001 vs. PSD group.
TMAO intensifies the dysregulation of neurotrophic mediators in the brain tissues of the PSD mouse model
The expression of two neurotrophic mediators (BDNF and GFAP) in the brain tissues of mice was further measured using the immunohistochemistry assay. Results revealed that the PSD mouse model had reduced BDNF and GFAP levels. Moreover, this phenomenon was aggravated by the introduction of TMAO in a dose-dependent way (Figures 3(a) and 3(b)). Thus, TMAO affected the metabolic balance of neurotrophic mediators. TMAO intensifies the dysregulation of neurotrophic mediators in brain tissues of PSD mice model. (a and b) The expression of neurotrophic mediators (BDNF and GFAP) in brain tissues of PSD mice model was analyzed by IHC assay. Scale bars for IHC: 50 μm. **p < .01 vs. Sham group, ***p < .001 vs. Sham group, ##p < .01 vs. PSD group, ###p < .001 vs. PSD group.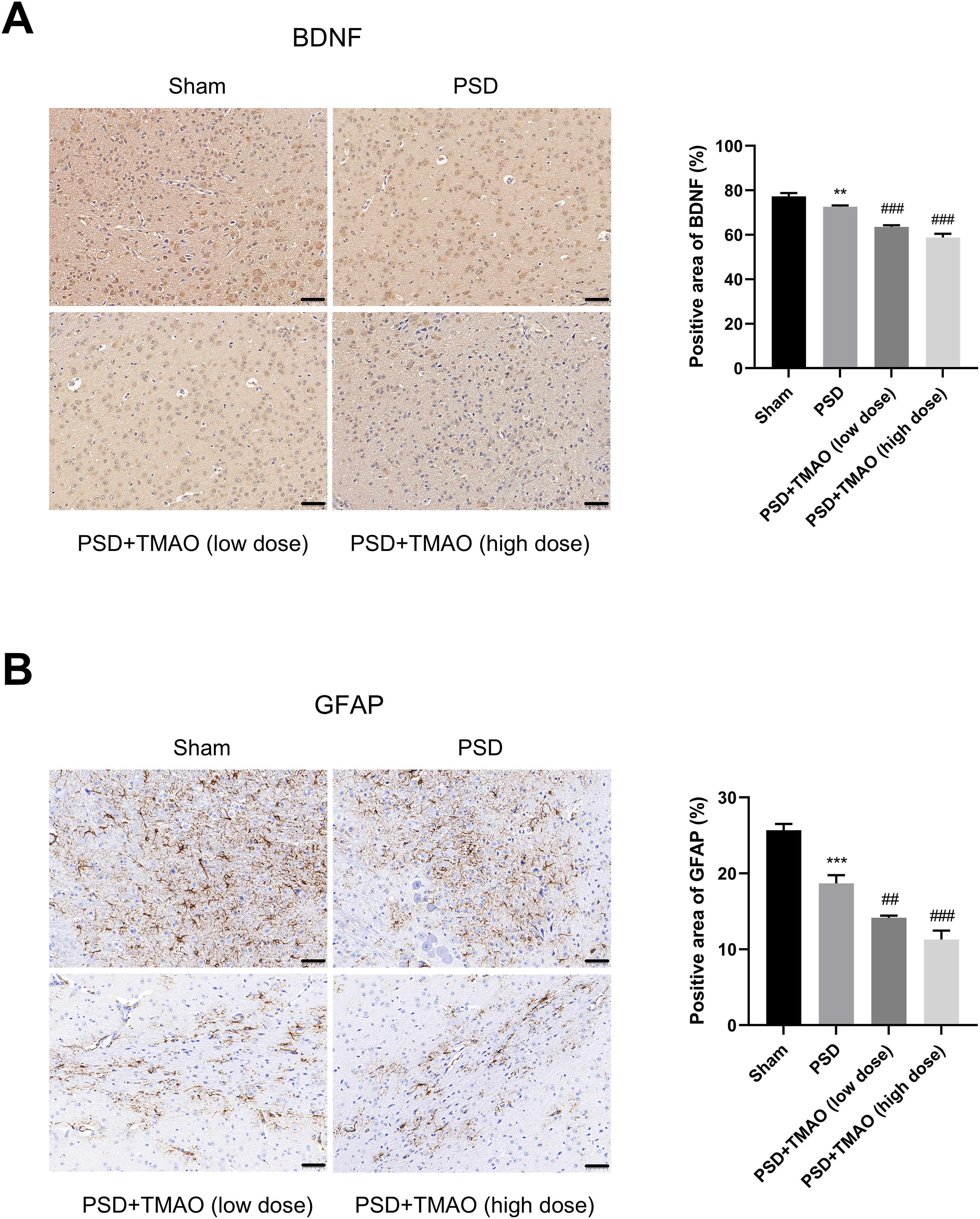
TMAO dose-dependently increases endothelial permeability and downregulates the abundance of tight junction proteins in hCMEC/D3 cells
TMAO reduced the abundance of tight junction proteins in the PSD mouse model in vivo. Therefore, the role of TMAO in brain vascular endothelial cells in vitro was further validated using the immortalized human brain microvascular endothelial cell line hCMEC/D3. The cytotoxicity of TMAO in hCMEC/D3 cells was initially analyzed, and results showed that TMAO did not have significant time- and dose-dependent cytotoxic effects on hCMEC/D3 cells (Figures 4(a) and 4(b)). The tight junction proteins of brain vascular endothelial cells maintained the integrity of the BBB and determined the endothelial permeability. Results showed that TMAO increased the endothelial permeability in a dose-dependent manner (Figure 4(c)). Moreover, TMAO dose-dependently reduced the mRNA and protein abundance of tight junction proteins (occludin and ZO-1) in hCMEC/D3 cells (Figures. 4(d)–4(h)). Overall, TMAO could downregulate the abundance of tight junction proteins and increase the permeability of brain microvascular endothelial cells in vitro. TMAO dose-dependently increases the permeability and downregulates the levels of tight junction proteins in hCMEC/D3 cells. (a) hCMEC/D3 cells were treated with gradient concentrations of TMAO for 24 h, and cell viability was measured by CCK8 assay. (b) hCMEC/D3 cells were exposed to 900 μM TMAO for 0, 4, 8, 12, 24, or 48 h, and CCK8 assay was employed to analyze cell viability. (c-h) hCMEC/D3 cells were treated with different doses of TMAO (0, 100, 300, 600, or 900 μM) for 24 h. (c) FITC-dextran permeability assay was conducted to assess the endothelial permeability in hCMEC/D3 cells. (d and e) RT-qPCR assay was implemented to determine the mRNA levels of occludin and ZO-1 in hCMEC/D3 cells. (f-h) Western blot assay was implemented to determine the protein levels of occludin and ZO-1 in hCMEC/D3 cells. *p < .05, **p < .01, ***p < .001.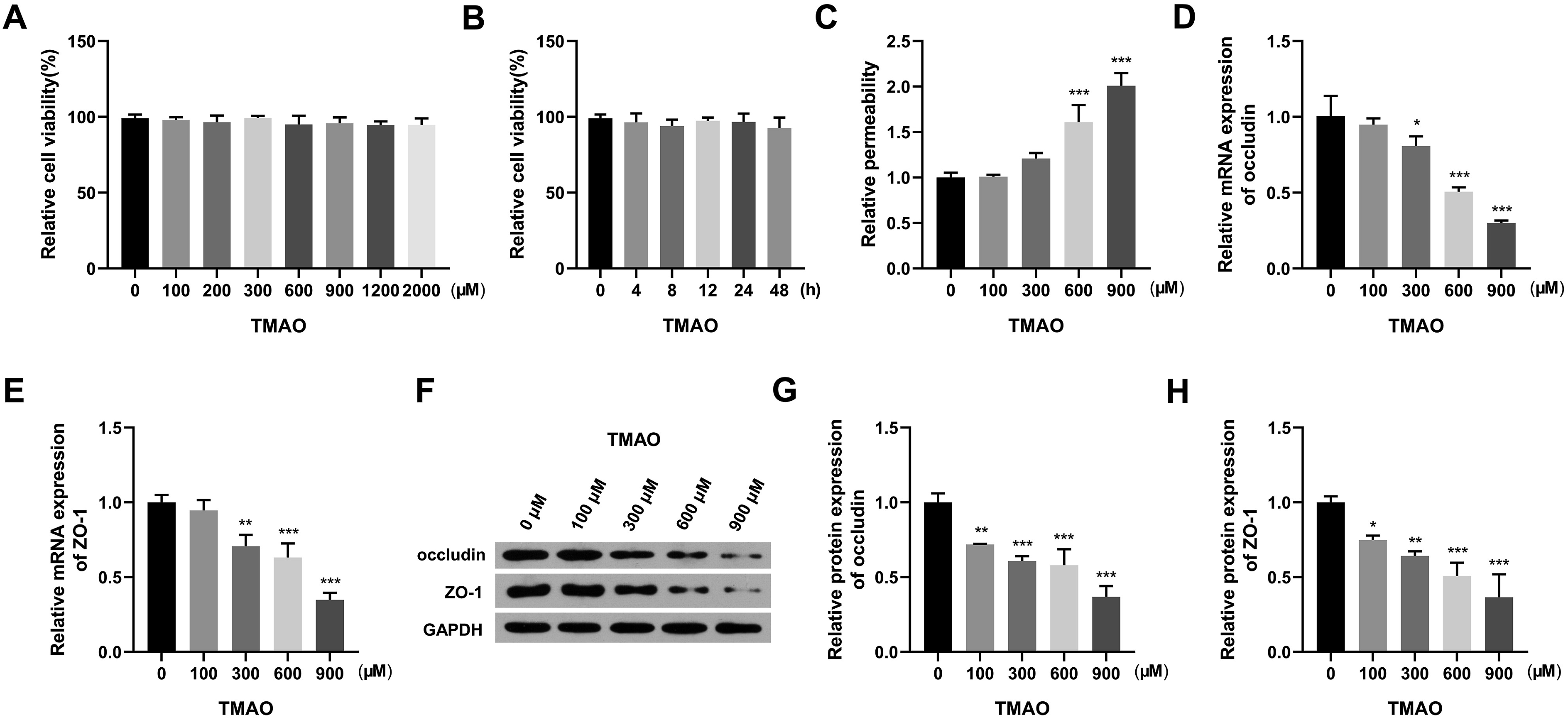
TMAO activates ROS-p38/mitogen-activated protein kinase signaling in hCMEC/D3 cells
We hypothesized that TMAO can downregulate the expression of tight junction proteins and endothelial permeability by activating ROS-p38/mitogen-activated protein kinase (MAPK) signaling. TMAO dose-dependently induced ROS fluorescence intensity in hCMEC/D3 cells (Figures 5(a) and 5(b)). In addition, TMAO treatment dose-dependently increased the ratio of p-p38/p38 (Figures 5(c) and 5(d)). Hence, TMAO activated ROS-p38/MAPK signaling in hCMEC/D3 cells. TMAO activates ROS-p38/MAPK signaling in hCMEC/D3 cells. (a-d) hCMEC/D3 cells were exposed to different doses of TMAO (0, 100, 300, 600, or 900 μM) for 24 h. (a and b) ROS generation was indicated by the fluorescence intensity. (c and d) The levels of total p38 and phosphorylated p38 in hCMEC/D3 cells were analyzed by western blot assay. *p < .05, **p < .01, ***p < .001.
The activation of ROS-p38/MAPK signaling is essential for TMAO-induced endothelial permeability in hCMEC/D3 cells
To explore whether the activation of ROS-p38/MAPK signaling was important for TMAO-mediated endothelial permeability, the p38/MAPK pathway inhibitor SB203580 was used to block this signaling pathway. Results revealed that TMAO-induced upregulation in endothelial permeability was prominently reversed by introducing SB203580 (Figures 6(a)). Further, TMAO-mediated reduction in the mRNA and protein abundance of occludin and ZO-1 was remarkably offset in the TMAO and SB203580 co-treatment groups (Figures. 6(b)–6(f)). Taken together, TMAO could increase the endothelial permeability by activating ROS-p38/MAPK signaling. The activation of ROS-p38/MAPK signaling is essential for TMAO-induced endothelial permeability in hCMEC/D3 cells. (a-f) hCMEC/D3 cells were divided into three groups: control (DMSO of the corresponding volume was added to the medium), TMAO (900 μM; 24 h), TMAO + SB203580 (2 μM, 24 h). (a) The endothelial permeability was analyzed by FITC-dextran permeability assay. (b and c) The mRNA levels of occludin and ZO-1 were detected by RT-qPCR. (d-f) The protein levels of occludin and ZO-1 were determined by western blot assay. *p < .05 vs. control group, **p < .01 vs. control group, ***p < .001 vs. control group, #p < .05 vs. TMAO group, ##p < .01 vs. TMAO group.
Discussion
The gut microbiota may influence cognition, mood, the development of anxiety and depression, and the central nervous system physiology via the microbiome–gut–brain axis.7,21,22 TMAO is a metabolite of the gut microbiota. Moreover, it was previously believed to be a useless nitrogen-containing waste. TMAO has been reported to play a role in the development of cardiovascular diseases, 23 neurological disorders, and depression. This study explored the potential connection between TMAO and PSD progression.
The MCAO animal model has been extensively used in the investigation of ischemic brain injury.24,25 Nevertheless, numerous studies have revealed the absence of inherent post-stroke depressive-like symptoms in mice. Conversely, individuals with hemiplegia after an ischemic stroke are prominently dependent on caregivers due to impaired mobility, which may lead to the development of depressive symptoms. To mimic the clinical course of PSD, the current study used an animal model that integrates the conventional stroke model (MCAO) with spatial constraint stress. 15 The TST and FST are extensively utilized to assess depression-like behaviors in rodent models.26–28 The dysfunction of cerebral vascular endothelial cells after cerebral ischemia results in BBB disruption and increased BBB permeability. This mechanism is related to a decreased abundance of tight junction proteins, ultimately leading to white matter atrophy and PSD development. The p38 signaling pathway is important in BBB disruption after acute intracerebral hemorrhage. Results revealed that the PSD group presented with a longer immobility time based on the TST and FST compared with the sham group. This finding was accompanied by reduced expressions of tight junction proteins and neurotrophic mediators (BDNF and GFAP) and activated p38 signaling pathway in brain tissues. BDNF is an essential indicator of depression. In addition, it plays an important role in neurogenesis promotion, damage repair, and neural plasticity.29,30 Previous studies have reported that a low BDNF level is related to the progression of depression-like behaviors in rodents.31–33 GFAP is the major intermediate filament protein in astrocytes, and a high GFAP level is a vital marker of astrocyte activation, which has protective effects on the nervous system.34,35 Therefore, low BDNF and GFAP levels in the brain tissues of the PSD group indicated neurological function deficits. The disturbance of the hypothalamic–pituitary–adrenal axis is a common symptom in patients with severe depression.36,37 The PSD group had higher serum cortisol level in comparison with the sham group, thereby indicating the involvement of the hypothalamic–pituitary–adrenal axis. This mouse model replicates several characteristics of human PSD. The introduction of TMAO dose-dependently intensified depression-like behaviors and changes in the expression of tight junction proteins and neurotrophic mediators in the PSD mice model.
To further validate the effect of TMAO in vitro, hCMEC/D3 cell line was utilized in this study. Results revealed that TMAO dose-dependently activated ROS-p38/MAPK signaling, decreased the expression of tight junction proteins, and increased the endothelial permeability in vitro. In addition, the role of the p38/MAPK pathway in this process was validated using the specific inhibitor SB203580. Moreover, the introduction of SB203580 prominently reversed TMAO-mediated effects in hCMEC/D3 cells. Therefore, TMAO downregulated the abundance of tight junction proteins and enhanced endothelial permeability by activating p38/MAPK signaling.
While the study provides valuable insights into the role of TMAO in PSD, several limitations should be acknowledged. Firstly, the variability in the gut microbiome composition between individual mice might influenced the observed effects of TMAO, as gut microbiota can modulate TMAO level and its impact on host metabolism and behavior. Moreover, the study predominantly relied on animal model, and the applicability of these results to human PSD requires cautious interpretation. Further investigation into the specific microbial taxa contributing to TMAO production and its pathological effects would provide deeper insights into the microbiome’s role in PSD. Addressing these limitations in future studies will be essential for translating these findings into therapeutic interventions.
Conclusion
TMAO reduced the expression of tight junction proteins and increased BBB permeability after stroke by activating the ROS-p38/MAPK pathway, thereby leading to the decrease of neurotrophic mediators, which eventually causes PSD. Therefore, it is important to identify the function and molecular mechanisms of TMAO in PSD.
Supplemental Material
Supplemental Material - Trimethylamine-N oxide enhances post-stroke depression progression via ROS-p38/MAPK signaling
Supplemental material for Trimethylamine-N oxide enhances post-stroke depression progression via ROS-p38/MAPK signaling by Yikui Hu, Yujie Liu, Hua Wang and Xun Wang in Human & Experimental Toxicology
Footnotes
Appendix
Acknowledgments
The author gratefully acknowledge the help of Scientific research project of Wuhan Health Commission (NO. WX20C40) and Surface Project of Natural Science Foundation of Hubei Province (NO. 2017CFB565) that funded this research.
Authors’ contributions
Yikui Hu and Yujie Liu designed and performed the experiments, wrote the manuscript. Hua Wang conducted the experiments. Xun Wang revised the manuscript. All authors have read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from Scientific research project of Wuhan Health Commission (NO. WX20C40) and Surface Project of Natural Science Foundation of Hubei Province (NO. 2017CFB565).
Ethical statement
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.