
Abstract
Aberrant mechanical forces were considered as an important factor for osteoarthritis (OA) pathogenesis. Plant homeodomain finger-containing protein 8 (PHF8) participated in osteogenic differentiation and inflammatory progression. However, the role of PHF8 in aberrant force-related OA remains to be elucidated. In this study, a fluid shear stress (FSS) model in ATDC5 cells and an anterior cruciate ligament transection (ACLT) animal model were constructed. The results revealed the decrease of PHF8 in aberrant force-induced cartilage damage in vitro and in vivo. PHF8 overexpression alleviated the aberrant force-induced cell apoptosis, extracellular matrix degradation, and inflammation. Chromatin immunoprecipitation (ChIP) assays demonstrated that PHF8 epigenetically regulated WWP2 expression through demethylating H3K9me2 at WWP2 promoter, which was influenced by FSS treatment. C-X-C chemokine receptor type 4 (CXCR4) was identified as a potential substrate of WWP2. Co-immunoprecipitation (Co-IP) and ubiquitination experiments further demonstrated WWP2 decreased the stability of CXCR4 via the ubiquitination pathway. Subsequently, rescue experiments validated reintroduction of WWP2 significantly attenuated the effects of PHF8 deletion on FSS-induced chondrocyte injury, and CXCR4 overexpression reversed the protective effects of WWP2 overexpression on chondrocyte injury in FSS-treated ATDC5 cells. Moreover, delivery of a PHF8 adeno-associated virus (AAV) into articular cartilage remarkably ameliorated the breakdown of cartilage matrix by ACLT in mice. In conclusion, our findings highlighted the importance of PHF8/WWP2/CXCR4 signaling pathway in aberrant force-induced cartilage injury, which might provide a novel insight on future epigenetic-based treatment of posttraumatic OA.
Introduction
Osteoarthritis (OA), characterized by the structural damage and functional failure of articular cartilage. 1 Articular cartilage is closely related to physical activity and often subjected to various external forces including stress, strain and pressure. 2 Whereas, in some cases, abnormal mechanical stress can result in the damage of cartilage tissue, cell apoptosis, degradation of extracellular matrix, as well as cartilage degradation. 3 Joint injuries, such as anterior cruciate ligament tears (ACLT) or damage during meniscus integrity loss, significantly increase the risk of post-traumatic osteoarthritis. Therefore, abnormal mechanical stress is considered as a key factor in OA progression, and elucidating its underlying pathogenesis is essential for the development of therapeutic strategies to alleviate posttraumatic OA.
Recently, emerging studies had revealed that epigenetic modifications including DNA methylation, histone modification, and no-coding RNAs was critical regulators for OA pathogenesis. 4 Among them, histone modifications, such as methylation, acetylation and ubiquitination, could control promoter activities for gene transcription and regulated the formation and maintenance of joints through epigenetic control. 5 For example, histone demethylase UTX influenced tri-methylation at lysine 27 of histone H3 (H3K27me3)-binding epigenomic landscapes, leading to articular chondrocyte anabolism and aggravating osteoarthritic degeneration. 6 H3K9 methylation repressed SOX9 transcription, slowing chondrogenesis and skeletal morphogenesis in mice deficient in AT-rich interactive domain 5b. 7 Histone lysine demethylase 7 A (KDM7A) was involved in regulation of osteoblast and osteoclast differentiation. 8 The X-linked di- and trimethylated lysine four of histone H3 (H3K4me2/3) demethylase KDM5C controlled female bone mass by promoting energy metabolism in osteoclasts. 9 Lian et al. reported that suppression of histone lysine demethylase 6A (KDM6A) elevated chondrocytic activity and alleviated OA development through inhibiting H3K27me3 enhancement of Wnt10a. 10
Plant homeodomain finger-containing protein 8 (PHF8), also known as KDM7B, contains a plant homeodomain (PHD) and a catalytic jumonji C domain. PHF8, a histone demethylase, modulated gene expression through removing epigenetic methyl markers (H3K9me2/1, H3K27me2, and H4K20me1) from the active promoter transcription start site (TSS) region, thus participating in the regulation of multiple biological processes. Recently, PHF8 was demonstrated to play a vital role in regulating bone development. Researchers had reported that PHF8 enhanced the osteogenic differentiation of marrow mesenchymal stem cells (BMSCs) in mice and promoted craniofacial bone repair. 11 The results from Liu et al. suggested that the increased PHF8 were observed during the osteogenic differentiation of periodontal ligament cells (PDLCs). 12 Additionally, PHF8 had been reported to participate in and regulate inflammatory response, and inflammation environment could lead to PHF8 loss. 13 More importantly, PHF8 promoted the osteogenic differentiation of rat BMSCs in osteoporosis rats through the Wnt/β-catenin pathway. 14 Osteogenic differentiation of BMSCs is essential for bone healing after joint injury. During the bone healing, the decreased capacity of BMSCs osteogenic differentiation could result in defects in bone formation, delay bone healing, and predispose patients to complications such as traumatic osteoarthritis. Based on the above findings, we speculated that PHF8 may be involved in the pathological process of OA. However, the biological function of PHF8 in aberrant mechanical forces-induced chondrocyte injury remains to be delineated.
Thus, we validated the functions of PHF8 in abnormal force-related cartilage injury as well as the underlying molecular mechanisms by using in vivo ACLT model and aberrant fluid shear stress (FSS)-treated chondrocytes.
Materials and methods
Human cartilage
The degenerated cartilage tissues were collected from 10 patients with a history of OA after total knee replacement surgery, and the normal cartilage tissues were collected from 10 individuals with the femoral neck fracture with no history of OA. The present study was approved by the Ethics Committee of Beijing Shijingshan Hospital (No. 20221754). All participants signed the informed consent.
Cell culture and fluid shear stress (FSS) treatment
ATDC5 cells were obtained from Shanghai Institute of Biosciences Cell Resource Center. Cells were maintained in Dulbecco’s modified eagle medium/Ham Nutrient Mixture F12 (DMEM/F12) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37°C within 5% CO2. The fluid shear stress was performed using a Flexcell FX-4000 strain unit (Flexcell, FX4000, Burlington, Ontario, Canada), and the loading force was 24 dyn/cm2 to stimulate cells for 1, 2 and 4 h. Cells in the control group were not subjected to FSS treatment. In brief, FSS treatment as conducted as described by previous study. 15 Chondrocytes were seeded onto a culture plate at a density of 2 × 105 cells and placed into a parallel-plate flow system that is a six-compartment laminar flow unit consisting six 75 × 25 × 1 mm culture plates. After 24 dyne/cm2 FSS treatment for 1, 2 or 4 h, the cells were collected for further experiments.
Cell transfection
ShRNA targeting PHF8 (PHF8 shRNA) and the corresponding control (NC shRNA) were obtained from GenePharma (Shanghai, China). Small interfering RNAs against WWP2 (WWP2 siRNA) and the negative control (NC siRNA) were provided by Thermo Fisher Scientific (Waltham, MA, USA). The gene-overexpression vectors (Ad-PHF8, Ad-WWP2 and Ad-CXCR4) and the control vector were purchased from GenScript Biotech Corp. (Nanjing, China). Plasmids and oligonucleotides transfection were performed with Lipofectamine 2000 transfection reagent under the suggestion of the manufacturer.
Osteoarthritis animal models
12-week-old C57BL/6 mice were provided by Shanghai Laboratory Animal Research Center (Shanghai, China). ACLT surgery was performed to establish an OA model as described by the previous study. 16 A total of 30 mice were randomized into the following five groups (6 mice in each group): Sham group, ACLT group, Sham + Vector group, ACLT + Vector group, and ACLT + AAV-PHF8 group. Prior to ACLT surgery, the mice were anesthetized with intravenous injection of 1% pentobarbitone (40 mg/kg). The left knees were exposed to medial parapatellar arthrotomy and ACLT after anesthetization. After that, the joint capsule and skin were sutured and closed. The control group (Sham group) received sham operation involving parapatellar incision in the right knee joint to expose the ACL. PHF8 Adeno-associated virus (AAV-PHF8) vectors were created and packaged by HanBio (Shanghai, China). One week after operation, vector AAV or AAV-PHF8 was delivered into the keen joints (1 × 1012 vg/mL, 10 μL). Eight weeks later, mice were sacrificed and joint tissues were collected for further analysis.
Safranin O fast green staining and immunohistochemistry (IHC) assay
The collected tissues were fixed in 4% paraformaldehyde for 24 h and decalcified. Demineralized samples were embedded in paraffin after gradient dehydration and cut into 5 μm thick sections for safranin O staining as previously indicated. 17
For immunohistochemistry, the sections were cultured with the primary antibodies at 4°C overnight: PHF8 (1:200; ab280887, Abcam), type II collagen (COLII; 1:200; ab307674, Abcam), A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-5 (ADAMTS-5; 1:200; ab231595, Abcam), Aggrecan (1:2000; ab313636, Abcam), and matrix metalloprotease 13 (MMP13; 1:100; ab315267, Abcam). Then, the slides were incubated with secondary antibodies for 1 h, stained with hematoxylin, dehydrated (ethanol), and mounted. Microscopy (BX53, Olympus, Japan) was used to capture the images.
Immunofluorescence assay
After being subjected to FSS treatment at 24 dyne/cm2 for 1 h, 2 h or 4 h, cells were fixed, permeabilized, and then blocked with 10% goat serum albumin for 30 min. Subsequently, cells were incubated with primary antibody against PHF8 (1:500; NBP3-21826, Novus) overnight at 4°C. Samples were washed with PBST and incubated with secondary antibody for 1 h at 37°C in the darkness. Next, the samples were stained with DAPI (10 nM) for 5 min. An inverted fluorescence microscope was used for observation (ZEISS, Germany).
RNA extraction and RT-qPCR analysis
Total RNA from samples and cultural cells were extracted by using TRIzol reagent (Takara, Japan). Reverse transcription was implemented with the PrimeScriptTM RT Reagent Kit (Takara Biotechnology, Dalian, China) to synthesize single-stranded cDNA. RT-qPCR was performed using a Power SYBR Green PCR kit (Takara Biotechnology, Dalian, China) with a 7500 Fast real-time PCR system (Applied Biosystems, USA). All the primers were obtained from Sangon Biotech (Shanghai) and the detailed sequences were listed in Table S1. GAPDH was used as a reference control using the 2 −ΔΔCT method.
Western blotting and co-immunoprecipitation (co-IP)
Cells or samples underwent lysis with Radio Immunoprecipitation Assay (RIPA) lysis buffer (Beyotime, Shanghai, China) supplemented with protease inhibitors. The BCA kit (Takara Biotechnology, Dalian, China) was applied for quantifying protein concentrations. After that, protein sample were separated by 10% SDS-PAGE, transferred to PVDF membranes, and blocked with 5% no-fat milk at room temperature for 1 h. Antibodies against PHF8 (1:1000; ab280887, Abcam), COLII (1:1000; ab307674, Abcam), ADAMTS-5 (1:2000; ab231595, Abcam), Aggrecan (1:1000; ab313636, Abcam), and MMP13 (1:1000; ab315267, Abcam), WWP2 (1:1000, 12197-1-AP, Proteintech), CXCR4 (1:100; ab124824, Abcam), Flag (1:50; ab205606, Abcam), HA (1:50; #3724, Cell Signaling Technology), and GAPDH (1:1000; ab8245, Abcam) were cultured with PVDF membranes at 4°C overnight. After incubating with secondary antibody for 1 h, the ECL-Plus Western Blot Analysis Detection System was used to observe the protein signals, and the relative protein band was analyzed by Image J software (NIH, Bethesda, MD, USA).
For immunoprecipitation, the cell lysates were incubated with the indicated antibodies in immunoprecipitation buffer for 4 h. Overnight incubation at 4°C allowed complexes to form, Protein A/G magnetic beads (B23202; Biotool) were added at 4°C for 3 h. Finally, immunoprecipitates were washed four times in RIPA buffer and analyzed by Western blot.
Ubiquitination assay
Flag-tagged CXCR4 and HA-UB were overexpressed in cells with WWP2 overexpression or WWP2 knockdown, followed by the treatment with MG132. Cells were lysed, boiled (10 min) and then sonicated to dissolve. Subsequently, the cell lysates were incubated with anti-CXCR4 antibody and protein A/G immunoprecipitation magnetic beads or anti-Flag magnetic beads for 12 h at 4°C. Analysis of CXCR4 ubiquitination was determined by immunoblotting with anti-HA antibodies.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was conducted using the EZ-ChIPTM kit (Millipore, Billerica, MA, USA). Generally, ATDC5 cells were cross-linked with 4% formaldehyde for 30 min before being quenched with glycine at room temperature. The lysates were treated with Buffer A, Buffer B, Micrococcal Nuclease (10011; CST), and ChIP buffer in this sequence to extract fragmented chromatins. Then, 10% volume of the lysates was used as the input control, and the remaining 90% lysate was immunoprecipitated with anti-PHF8 (ab280887; Abcam), anti-H3K4me3 (ab8580; Abcam), anti-H3K9me1 (ab176880; Abcam), anti-H3K9me2 (ab1220; Abcam), anti-H4K20me1 (ab177188; Abcam), or anti-IgG (ab37415; Abcam) antibodies. The mixtures of protein-DNA and antibody were subsequently incubated with Protein G Agarose Beads. Then the complex containing the magnetic beads was collected, and the DNA in the complex was eluted with elution buffer (1% SDS and 0.1 M NaHCO3) at 37°C for 30 min. Finally, immunoprecipitated DNA and input were purified, and WWP2 enrichment in the immunoprecipitated complex was detected by RT-qPCR analysis. The results are displayed as the percentage of immunoprecipitated over input values (% input).
Cell viability assay
Cell viability of indicated ATDC5 cells was evaluated using the CCK-8 kit (Takara). Briefly, cells were incubated in a 96-well plate (1 × 104 cells/well) for 48 h. After indicated treatment according to the experimental grouping, 10 μL of CCK-8 solution was added to each well for additional 2 h culture. The absorbance value at 450 nm wavelength was detected using a microplate reader (BioTek, Biotek Winooski, Vermont, USA).
Cell apoptosis analysis
Apoptotic cells were determined by using the Annexin V-FITC/PI apoptosis detection kit (Beijing Biosea Biotechnology, Beijing, China). Briefly, ATDC5 cells were seeded into 6-well plate (3 × 104 cells/well) and subjected to different treatment or transfection. After 48 hours of culture, the treated ATDC5 cells were re-suspended in buffer and then stained with 10 μL Annexin V-FITC/PI solution in the dark at 37°C. Finally, the stained cells were measured by flow cytometry (BD Biosciences, San Jose, CA, USA), and the FlowJo software (Treestar, Ashland, OR, USA) was used for analysis.
Enzyme linked immunosorbent assay (ELISA)
The levels of tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6) and IL-1beta (IL-1β) were measured by using the ELISA kits (Abcam, Cambridge, MA) as per manufacturer’s instructions. For in serum in vivo, blood samples were collected from the abdominal aorta, and then centrifuged. For in vitro assays, removing the cells and other debris via centrifugation to collect the supernatant culture after the indicated treatment. Finally, the absorbance was assessed at 450 nm within 30 min.
Statistical analysis
The data were expressed as mean ± SEM. Statistical analysis between different groups were conducted using SPSS 18.0. Student’s t test was used for the comparison between two groups. Comparison among multiple groups was analyzed using one-way analysis of variance (ANOVA) or Two-way ANOVA. p < .05 was considered statistically significant.
Results
Aberrant force decreased the levels of PHF8 expression in vitro and in vivo
In this study, ATDC5 cells were exposed to FSS (24 dyne/cm2) treatment for 1, 2 and 4 h to induce chondrocytes injury. We observed that the levels of PHF8 mRNA and protein expression were significantly decreased in chondrocytes treated with FSS using RT-qPCR analysis and Western blotting (Figure 1(a) and (b)). Consistently, immunofluorescence assay results showed an obvious decrease of the staining intensity of PHF8 in FSS-treated chondrocytes (Figure 1(c)). Then, ACLT mice model was established for in vivo investigation. Immunohistochemistry, RT-qPCR and Western blotting experiments showed PHF8 level was significantly decreased in ACLT group compared to the Sham group (Figure 1(d)–(f)). Then the mRNA and protein levels of PHF8 expression in cartilage tissues from the OA group and normal group were detected, and the decreased levels of PHF8 were observed both at mRNA and protein levels (Figure 1(g) and (h)). Taken together, our data suggested that PHF8 may play an important role during OA pathogenesis. Aberrant force decreased the levels of PHF8 expression in vitro and in vivo. (a) and (b). ATDC5 cells were subjected to 24 dyne/cm2 FSS for 1, 2 or 4 h. Relative expression levels of PHF8 mRNA and protein in FSS-treated chondrocytes were detected by RT-qPCR and Western blotting, respectively. (c). Immunofluorescent assay was used to detect the staining intensity of PHF8 in FSS-treated chondrocytes. Magnification: ×1000. (d). Immunohistochemistry staining of PHF8 in joint samples from mice subjected to sham and ACLT surgery. Scale bar: 250 μm. (e) and (f). The relative mRNA and protein levels of PHF8 in tissue samples from sham and ACLT group (6 mice in each group). (g). The cartilage tissues in OA group were obtained from the patients with a history of OA after total knee replacement surgery, and the normal cartilage tissues were collected from the volunteers with the femoral neck fracture. Relative expression levels of PHF8 in cartilage tissues obtained from normal group and OA group was determined by RT-qPCR. (h). Relative expression levels of PHF8 in cartilage tissues obtained from normal group and OA group was determined by Western blotting. Data are shown as means ± SEM, n = 3 in (a-c); n = 6 in (e, f); n = 10 in (g, h). Student’s t test and One-way ANOVA was used for the comparison in this study. *p < .05. * *p < .01.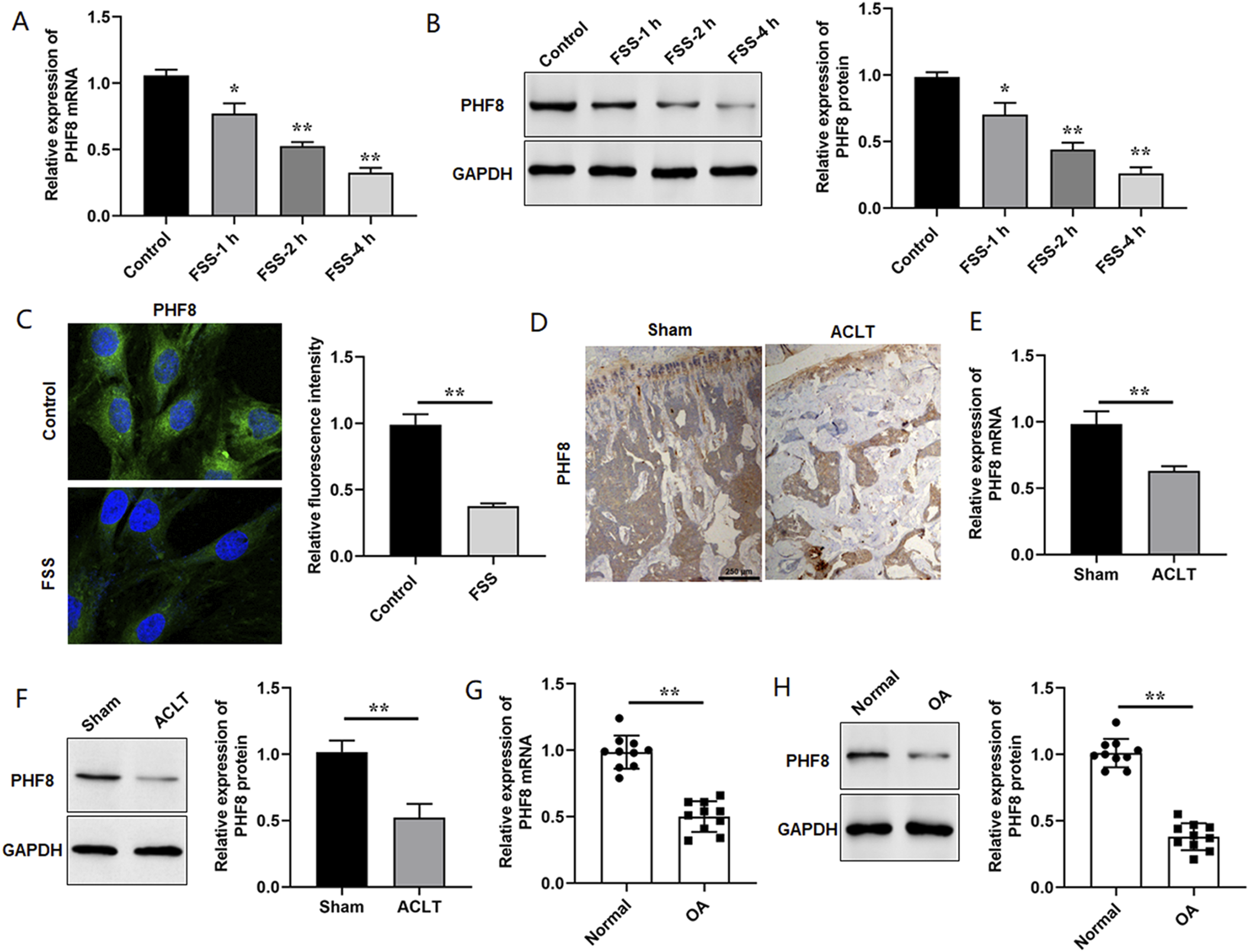
Overexpression of PHF8 restrained FSS-induced cell apoptosis, extracellular matrix degradation, and inflammation
Ad-PHF8 or PHF8 shRNA was transfected into ATDC5 cells to overexpress or silence PHF8, and the PHF8 expression was confirmed by RT-qPCR and Western blot analysis (Figure 2(a) and (b)). Then, ATDC5 cells were transfected with Ad-PHF8 or Vector, followed by subjecting to 24 dyne/cm2 FSS for 2 h. First, we found that the decrease of PHF8 induced by FSS treatment was reversed by PHF8 overexpression (Figure 2(c)). Further experiments suggested that FSS promoted chondrocyte apoptosis, while PHF8 overexpression prevented FSS-induced cell apoptosis (Figure 2(d)). CCK-8 assay results indicated the decreased cell viability of ATDC5 cells induced by FSS treatment was significantly weakened by PHF8 overexpression (Figure 2(e)). Furthermore, PHF8 overexpression improved the expression of cartilage-related markers in ATDC5 cells and rescued the destructive functions of aberrant FSS (Figure 2(f) and (g)). Then, we detected the concentrations of inflammatory cytokines in different groups, and found that PHF8 overexpression notably alleviated decreased the levles of TNF-α, IL-1β and IL-6 in FSS-treated ATDC5 cells (Figure 2(h)). Overall, the above data showed that PHF8 overexpression ameliorated inflammation response and chondrocyte damage induced by FSS treatment, which suggested the important role of PHF8 in aberrant force-related OA. PHF8 overexpression alleviated FSS-induced chondrocyte injury. (a) and (b). ATDC5 cells were transfected with Ad-PHF8, PHF8 shRNA or their negative controls, followed by the RT-qPCR and Western blot analysis. Relative expression levels of PHF8 mRNA and protein were detected. (c). After transfected with Ad-PHF8 or Vector, ATDC5 cells were subjected to 24 dyne/cm2 FSS for 2 h. Relative expression levels of PHF8 protein were detected by Western blotting. (d). Apoptotic cells were determined by flow cytometry analysis. (e). Cell viability was detected by CCK-8 assay. (f) and (g). Relative expression of COLII, Aggrecan, ADAMTS-5, and MMP13 at mRNA and protein levels was detected by RT-qPCR and Western blotting, respectively. (h). Concentrations of pro-inflammatory cytokines (TNF-α, IL-1β and IL-6) in the culture supernatant were detected by ELISA kits. Data are shown as means ± SEM, n = 3 in (A- H). One-way ANOVA was used for the comparison in this study. *p < .05. * *p < .01.
PHF8 epigenetically regulated WWP2 expression via demethylating H3K9me2 at WWP2 promoter
It is reported that PHF8 could serve as a transcription coactivator to epigenetically regulate the target expression.
18
To further investigate the regulatory mechanisms of PHF8 in FSS-induced chondrocyte injury, we found that the promoter of WWP2 has the potential binding sites of PHF8 using hTFtarget online software. Thus, we determined the expression levels of WWP2 in chondrocytes exposed to 24 dyne/cm2 FSS for 1, 2 or 4 h. The results showed that WWP2 was obviously decreased in FSS-treated ATDC5 cells (Figure 3(a) and (b)). We next performed ChIP assays to verify the binding of PHF8 with the WWP2 promoter. The results revealed that PHF8 could bind to the WWP2 promoter, which was weakened by FSS treatment (Figure 3(c)). The facts that PHF8 overexpression promoted the binding of PHF8 at the gene body regions supported the specificity of PHF8 binding to the promoter of WWP2 (Figure 3(c)). Furthermore, ChIP assays with indicated antibodies against different histone modifications (H3K4me3, H3K9me1/2 and H4K20me1) were performed for WWP2 gene. Without FSS treatment, reintroduction of PHF8 reduced the H3K9me2 levels at the WWP2 promoter, but it did not affect the levels of H3K4me3, H3K9me1 and H3K20me1 (Figure 3(d)). After FSS treatment, H3K4me3 levels were significantly decreased, which supported the recruitment of PHF8 as it binds to this methylated histone via its PhD domain. We also observed that overexpression of PHF8 increased the H3K4me3 level but decreased the levels of H3K9me2 at the WWP2 promoter (Figure 3(d)). Finally, RT-qPCR and Western blot analysis were conducted to detect the expression levels of WWP2, and we found that the decrease of WWP2 induced by FSS treatment was reversed by PHF8 overexpression (Figure 3(e) and (f)). In summary, our results suggested that PHF8 epigenetically regulated WWP2 expression via demethylating H3K9me2 at WWP2 promoter, which was influenced by FSS treatment. PHF8 epigenetically regulated WWP2 expression via demethylating H3K9me2 at WWP2 promoter. (a) and (b). RT-qPCR and Western blot analysis were performed to determine the expression levels of WWP2 mRNA and protein in chondrocytes exposed to 24 dyne/cm2 FSS for 1, 2 and 4 h. (c). Cells were transfected with Vector or Ad-WWP2 followed by the stimulation with FSS. The binding of PHF8 with WWP2 promoter was detected by CHIP assay. (d). Similar CHIP assays as (b) with indicated antibodies against different histone modifications were performed for WWP2 gene. (e) and (f). After transfected with Ad-PHF8 or Vector, ATDC5 cells were subjected to 24 dyne/cm2 FSS for 2 h. Relative expression levels of WWP2 mRNA and protein were detected by RT-qPCR and Western blotting, respectively. Data are shown as means ± SEM, n = 3 in ((a)-(f)). One-way ANOVA was used for the comparison in this study. *p < .05. * *p < .01.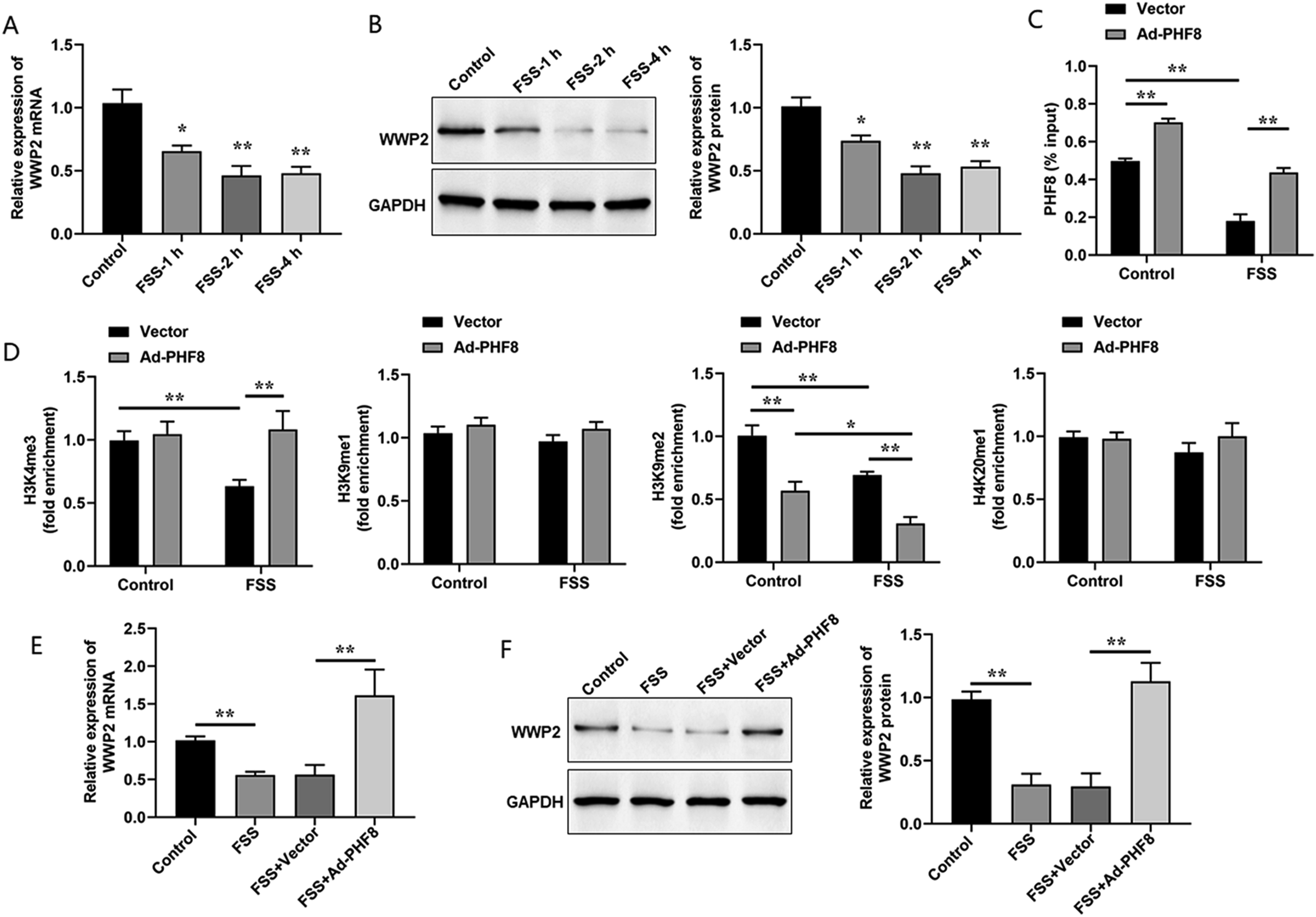
Overexpression of WWP2 attenuated the effects of PHF8 deletion on FSS-induced chondrocyte injury
Then, whether PHF8 regulated FSS-induced chondrocyte injury through WWP2 was investigated. Ad-WWP2 or WWP2 shRNA was transfected into chondrocytes (Figure 4(a) and (b)). Subsequently, the rescue assays were conducted by transfecting PHF8 shRNA alone or together with Ad-WWP2 into ATDC5 cells followed by FSS treatment, and the expression levels of WWP2 protein were determined (Figure 4(c)). Upon FSS treatment, PHF8 knockdown significantly decreased the cell viability of ATDC5 cells, which was weakened by overexpression of WWP2 (Figure 4(d)). Further experiments suggested that chondrocyte apoptosis was obviously increased by PHF8 knockdown in FSS-treated cells, while WWP2 overexpression reversed these results (Figure 4(e)). Furthermore, we observed that knockdown of PHF8 decreased the expression levels of COLII and Aggrecan, but increased the expression levels of ADAMTS-5 and MMP13 in FSS-treated ATDC5 cells, thus further aggravating the destructive functions of aberrant forces (Figure 4(f) and (g)). Yet, reintroduction of WWP2 alleviated the effects of PHF8 knockdown on the expression of cartilage-related markers in FSS-treated ATDC5 cells (Figure 4(f) and (g)). Then, the results also illustrated that PHF8 knockdown remarkably further facilitated inflammation response in FSS-treated ATDC5 cells, as evidenced by the increase of TNF-α, IL-1β and IL-6 levels, while WWP2 overexpression reversed these results (Figure 4(h)). These results demonstrated that FSS-induced chondrocyte injury was depend on the PHF8/WWP2 axis. Overexpression of WWP2 attenuated the effects of PHF8 deletion on FSS-induced chondrocyte injury. (a) and (b). ATDC5 cells were transfected with Ad-WWP2, WWP2 shRNA or their negative controls, followed by RT-qPCR and Western blot analysis. Relative expression levels of WWP2 mRNA and protein were detected. (c). ATDC5 cells were transfected with PHF8 shRNA alone or together with Ad-WWP2, and then cells were subjected to 24 dyne/cm2 FSS for 2 h. Relative expression levels of WWP2 protein were detected by Western blotting. (d). Cell viability was detected by CCK-8 assay. E. Apoptotic cells were determined by flow cytometry analysis. (f) and (g). Relative expression levels of COLII, Aggrecan, ADAMTS-5, and MMP13 mRNA and protein were detected by RT-qPCR and Western blotting, respectively. (h). Concentrations of pro-inflammatory cytokines (TNF-α, IL-1β and IL-6) in the culture supernatant were detected by ELISA kits. Data are shown as means ± SEM, n = 3 in ((a)-(h)). One-way ANOVA was used for the comparison in this study. *p < .05. * *p < .01.
WWP2 combined directly with CXCR4, and degraded CXCR4 through the proteasome-dependent pathway
Upon Ubibrowser online software, we found that CXCR4 may serve as one of the substrates of ubiquitin protease WWP2. Co-immunoprecipitation of endogenous proteins was applied to verify the interaction between CXCR4 and WWP2 (Figure 5(a)). Co-immunoprecipitation of exogenous proteins was performed by using HA-tagged WWP2 and Flag-CXCR4 to determine the interaction between CXCR4 and WWP2 (Figure 5(b)). We further investigated whether FSS treatment affected the interaction between WWP2 and CXCR4. ATDC5 cells were treated with 24 dyne/cm2 FSS for 2 h or not, and the results showed that FSS treatment decreased binding between WWP2 and CXCR4 (Figure 5(c)). The above data indicated that WWP2 participated in FSS-induced chondrocyte injury through interacting with CXCR4. WWP2 combined directly with CXCR4, and degraded CXCR4 through the proteasome-dependent pathway. Co-immunoprecipitation was conducted to verify the interaction between WWP2 and CXCR4. Endogenous CXCR4 coimmunoprecipitated with endogenous WWP2 (a), as did HA-tagged WWP2 and Flag-CXCR4 (b). (c). Co-IP analysis of interaction between WWP2 and CXCR4 in ATDC5 cells with or without FSS treatment was showed. (d). Relative expression levels of CXCR4 protein in WWP2-overexpressed cells were determined by Western blotting. (e). Relative expression levels of CXCR4 protein in WWP2-silenced cells through transfecting with three target sequences of siRNA-WWP2 were determined by Western blotting. (f). Western blot analysis was used to demonstrate the impact of WWP2 knockdown on stabilizing CXCR4 protein levels at various times after CHX administration. (g). Western blot analysis was used to demonstrate the impact of WWP2 knockdown on stabilizing CXCR4 protein levels at various times after MG132 administration. H. Flag-CXCR4, HA-WWP2, and HA-Ub were co-expressed then treatment with MG132 or not. CXCR4 was isolated by IP, and CXCR4 ubiquitination levels were assessed with anti-HA antibodies. I. WWP2 siRNA, Flag-CXCR4 and HA-Ub were co-expressed and then treatment with MG132 or not. CXCR4 was isolated by IP, and CXCR4 ubiquitination levels were assessed with anti-HA antibodies. Data are shown as means ± SEM, n = 3 in ((f), (g)). Two-way ANOVA was used for the comparison in this study. *p < .05. * *p < .01.
Then, a gradient of HA-tagged WWP2 were transfected into cells to exogenously overexpress WWP2. The results validated that the expression levels of CXCR4 were gradually decreased with the increase of WWP2 expression (Figure 5(d)). Three WWP2-siRNA sequences were designed and transfected into ATDC5 cells to silence WWP2, and we found that these three sequences notably decreased WWP2 expression, but distinctly elevated CXCR4 levels (Figure 5(e)). To verify whether WWP2 decrease the expression levels of CXCR4 by suppressing the transcription of the CXCR4, ATDC5 cells were transfected with NC siRNA or WWP2 siRNA and then treated with transcription inhibitor cycloheximide (CHX) in time-gradient. We observed that CXCR4 protein was significantly decreased in the NC siRNA group, while no obvious changes were observed in the WWP2 siRNA group (Figure 5(f)), which indicated WWP2 showed no effects on CXCR4 transcription. Subsequently, ATDC5 cells were treated with MG132 in time-gradient after transfection with NC siRNA or WWP2 siRNA. The results showed that the expression levels of CXCR4 protein elevated over time after MG132 treatment in the NC siRNA group. Whereas, the expression levels of CXCR4 protein were maintained at higher levels after MG132 treatment in the WWP2 siRNA group (Figure 5(g)).
HA-ubiquitin, HA-WWP2 and Flag-CXCR4 were co-transfected into HEK 293T cells followed by the treatment of MG132, and then the levels of CXCR4 ubiquitination were detected. We found that the ubiquitination levels of CXCR4 was effectively increased by WWP2 overexpression (Figure 5(h)). Moreover, Flag-CXCR4 and HA-ubiquitin were overexpressed in WWP2 depletion cells, and we found that WWP2 knockdown obviously reduced the CXCR4 ubiquitination levels (Figure 5(i)). Therefore, our findings revealed that WWP2 interacted with CXCR4 and degraded CXCR4 via the deubiquitination pathway.
Reintroduction of CXCR4 attenuated the effects of WWP2 overexpression on FSS-induced chondrocyte injury
Then, we further verified whether WWP2 participated in FSS-induced chondrocyte injury through CXCR4. ATDC5 cells were transfected with Ad-WWP2 or Ad-CXCR4 alone, or Ad-WWP2 and Ad-CXCR4 together followed by the treatment with FSS, and the expression levels of CXCR4 protein were determined by Western blot analysis (Figure 6(a)). Overexpression of CXCR4 notably reduced chondrocyte cytotoxicity and facilitated chondrocyte apoptosis in FSS-induced cells (Figure 6(b) and (c)). And the effects of WWP2 overexpression on chondrocyte viability and apoptosis were notably attenuated by reintroduction of CXCR4 (Figure 6(b) and (c)). Moreover, CXCR4 overexpression significantly alleviated the protective effects of WWP2 overexpression on chondrocyte injury, as evidenced by decreasing COLII and Aggrecan expression, elevating ADAMTS-5 and MMP13 expression, and promoting inflammatory responses (Figure 6(d)–(f)). Herein, our data suggested that the regulatory functions of WWP2 in FSS-treated ATDC5 cells were achieved through CXCR4. Reintroduction of CXCR4 ameliorated the effects of WWP2 overexpression on FSS-induced chondrocyte injury. ATDC5 cells were transfected with Ad-WWP2 or Ad-CXCR4 alone, or both Ad-WWP2 and Ad-CXCR4, and then cells were subjected to 24 dyne/cm2 FSS for 2 h. (a). Relative expression levels of CXCR4 protein were detected by Western blotting. (b). Cell viability was detected by CCK-8 assay. (c). Apoptotic cells were determined by flow cytometry analysis. (d) and (e). Relative expression levels of COLII, Aggrecan, ADAMTS-5, and MMP13 mRNA and protein were detected by RT-qPCR and Western blotting, respectively. (f). Concentrations of pro-inflammatory cytokines (TNF-α, IL-1β and IL-6) in the culture supernatant were detected by ELISA kits. Data are shown as means ± SEM, n = 3 in ((a)-(f)). One-way ANOVA was used for the comparison in this study. *p < .05. * *p < .01.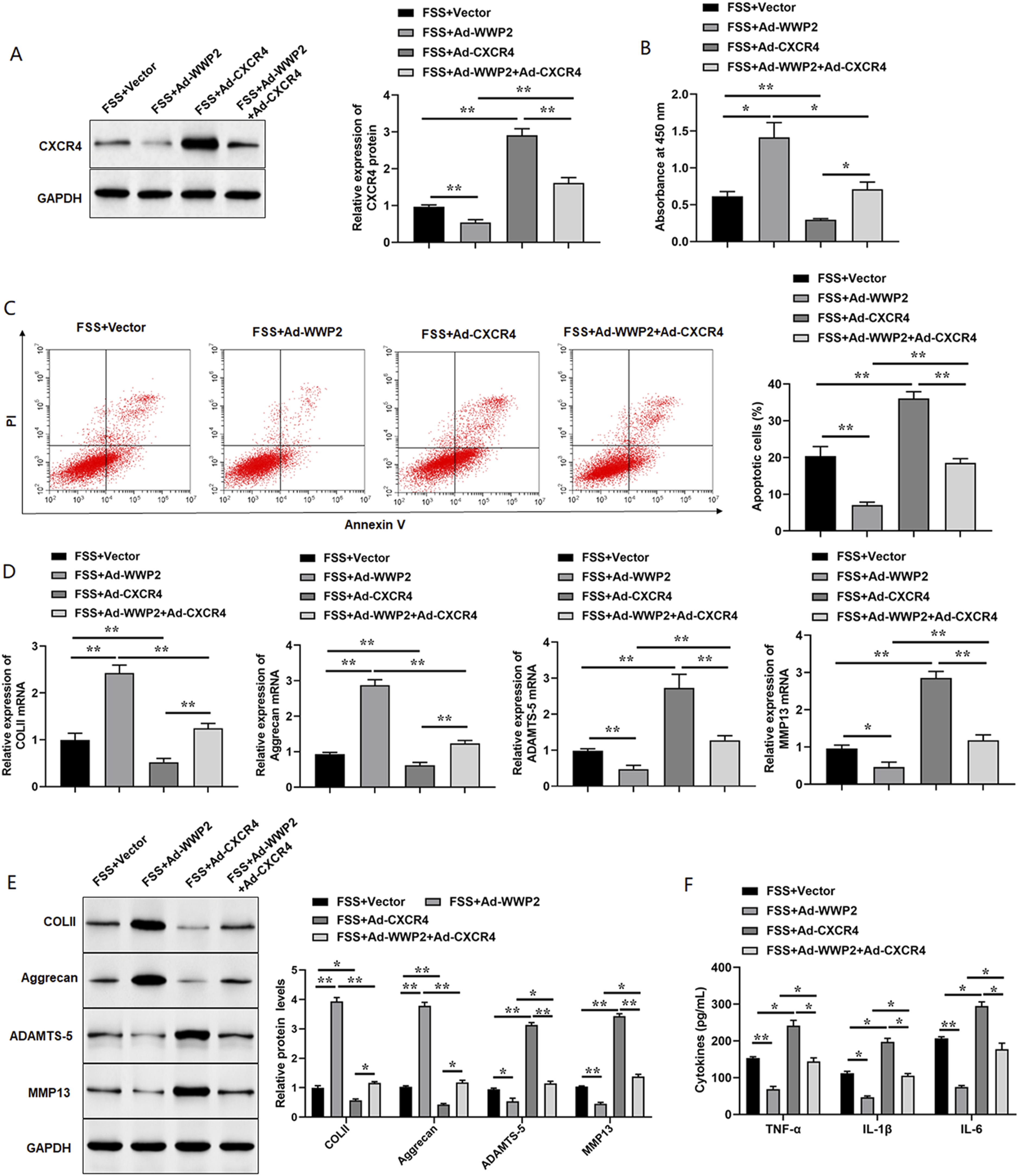
PHF8 effectively attenuated OA progression
Based on the abovementioned findings, we further confirmed the role of PHF8 regulation in mechanical-related OA pathogenesis in vivo. Using Safranin O fast green staining, we observed that safranin O-positive cells were distinctly elevated in the ACLT + Vector group compared with the ACLT + Sham group, indicating that ACLT-induced proteoglycan loss were rescued by AAV-PHF8 injection (Figure 7(a)). Western blot analysis also suggested that the changes in COLII, Aggrecan, ADAMTS-5 and MMP13 expression were weakened by PHF8 overexpression, indicating AAV-PHF8 could rescue the OA progression caused by ACLT (Figure 7(b) and (c)). Immunohistochemistry analyses in the three groups revealed that overexpression of PHF8 elevated the expression levels of WWP2 and reduced the expression levels of CXCR4, therefore inhibiting the OA progression caused by ACLT surgery (Figure 7(d)–(g)). Furthermore, AAV-PHF8 injection significantly alleviated inflammatory response after ACLT surgery, as evidenced by the decrease levels of TNF-α, IL-1β and IL-6 (Figure 7(h)–(j)). Collectively, these data illustrated that PHF8 are involved in ACLT-induced OA pathogenesis in vivo. PHF8 effectively attenuated OA progression. One week after sham or ACLT operation, a total of 10 μL (approximately 1 × 1012 vg/mL) of AAV vector, or PHF8-AAV was delivered into the knee joint (6 mice in each group). (a). Safranin-O-fast green staining of joint samples in different groups was shown. Scale bar: 100 μm. (b) and (c). Relative expression levels of MMP13, ADAMTS-5, COLII, and Aggrecan were detected by Western blotting. (d)-(g). Immunohistochemistry staining of PHF8, WWP2 and CXCR4 in tissue samples from mice with different treatments were showed. Scale bar: 250 μm. (h)-(j). The levels of pro-inflammatory cytokines (TNF-α, IL-1β and IL-6) were detected by ELISA kits. Data are shown as means ± SEM, n = 6 in ((c), and (e)-(j)). One-way ANOVA was used for the comparison in this study. *p < .05. * *p < .01.
Discussion
ACLT is considered as the most common knee injury and often resulted in damage to the meniscus, other ligaments, or cartilage of the joint. Joint damage caused by aberrant mechanical load is thought to contribute to OA development. Mechanical factors seriously affected the biosynthetic activity and ECM homeostasis of chondrocytes. 19 With or without ACL-reconstruction surgery, more than 50% of patients with ACL-injury patients may develop post-traumatic OA within 5-20 years of the injury. 20 Therefore, unlike other studies that induce OA using pro-inflammatory mediators such as IL-1 β or LPS, we applied aberrant mechanical stress to chondrocytes in vitro to simulate more physiologically-like conditions in the OA environment and established ACLT mouse models to simulate the occurrence and progression of OA in vivo. Researches had revealed that high shear stress caused inflammatory responses, cell apoptosis and death, and matrix degradation, thus leading to cartilage degradation and OA-like pathological features.21,22 Aberrant mechanical loading could cause cartilage degeneration through modulating the autophagy and apoptosis programs in chondrocytes. 23 In agreement with previous findings, our data suggested that high shear stress displayed the negative impact on chondrocytes, as evidenced by the increase of apoptotic cells, decreased levels of COLII and Aggrecan expression and increased levels of DAMTS-5 and MMP13 expression, as well as the increased levles of TNF-α, IL-1β and IL-6. Moreover, FSS treatment on primary chondrocytes induced cartilage degeneration and degradation. 15 In conclusion, aberrant high shear stress contributes to cartilage damage.
Recently, several studies had uncovered that epigenetic regulation was participated in biological activities or diseases which were related to mechanical forces. For example, histone methyltransferase enhancer of zeste homolog 2 (EZH2) was verified as a FSS-responsive gene, and the decrease in EZH2 under FSS restrained cell cycle, thus allowing endothelial cells to enter quiescence. 24 Zhang et al. validated that the mechanically sensitive suppressed the osteogenic differentiation of periodontal ligament stem cells through epigenetic pathways. 25 In addition, long term mechanical forces could affect the epigenetic modifications regulating osteogenic differentiation of human adipose tissue multipotential stromal cells (hAT-MSCs) and contribute to accelerated osteogenesis in vitro. 26 Epigenetic disorders had been reported to be closely related to the pathogenesis of OA. A study revealed that aberrant mechanical stress enhanced the binding of bromodomain-containing protein four to H3K27ac on the promoter region of triggering receptor expressed on myeloid cell 1 which was related to the stress and inflammatory pathways, thus leading to temporomandibular joint OA-like pathological changes. 27 Jin et al. reported that aberrant force-induced the upregulation of jumonji domain-containing protein-3 (JMJD3) demethylated H3K27me3 at the nuclear receptor subfamily four group A member 1 (NR4A1) promoter, and then lead to chondrocyte injury. 28 Together, these findings highlighted the critical role of epigenetic regulators in shear stress-related OA.
As a histone demethylase, PHF8 had been proved to regulate the inflammatory process and osteogenic differentiation by catalyzing the removal of histone sites H3K9 and H4K20. 29 A study reported that PHF8 demethylated H3K9me1 at the promoter region of special AT-rich sequence-binding protein 2, triggering BMSCs osteogenic differentiation and facilitating bone formation. 11 PHF8 was significantly downregulated in FSS-treated chondrocytes and ACLT mice and PHF8 overexpression improved the expression of cartilage-related markers in ATDC5 cells and rescued the destructive functions of aberrant force. In vivo experiments further verified the PHF8 effectively attenuated ACLT-induced OA progression, suggesting that PHF8 was involved in excessive mechanical stress-induced cartilage injury. Furthermore, the decreased PHF8 expression was found in osteoporosis rats, and PHF8 reintroduction elevated osteogenic differentiation in Rbmsc. 14 PHF8 mediated inflammatory processes 13 and regulated the osteogenic differentiation of PDLCs in inflammatory environments. 12 In agreement with previous findings, our results also demonstrated that overexpression of PHF8 notably alleviated inflammation response in FSS-treated ATDC5 cells and in ACLT-induced OA mice. All above findings indicated that PHF8 showed protective effects on osteoarthropathy serving as pharmacological candidates for the treatment of posttraumatic OA.
WWP2, a member of the NEDD4 family of E3 ubiquitin ligases, participated in various biological processes via ubiquitylation pathway. 30 WWP2 transcription in cartilage was epigenetically regulated and played a responsive role in the pathophysiological processes of OA.31,32 In addition, WWP2 was demonstrated to be a marker for hypertrophic chondrocytes in OA knee joints. 33 Thus, we detected the expression levels of WWP2 in chondrocytes exposed to FSS treatment, and found that WWP2 was significantly decreased in FSS-treated ATDC5 cells, suggesting WWP2 could be decreased by high shear stress. More interestingly, ChIP analysis confirmed that PHF8 epigenetically regulated WWP2 expression through demethylating H3K9me2 at WWP2 promoter, which was influenced by FSS treatment. The rescue experiments revealed that WWP2 overexpression notably alleviated the effects of PHF8 knockdown on cartilage-related markers expression, cell apoptosis, and the secretion of inflammatory cytokines, which was consistent with the findings of Mokuda 34 showing that WWP2 expression level was downregulated in human OA cartilage and lack of WWP2 expression exhibited aggravated spontaneous and surgically induced OA. To further elucidate the underlying mechanism of WWP2 in OA, we found that CXCR4 may serve as one of the substrates of ubiquitin protease WWP2 using Ubibrowser online software, and the expression levels of CXCR4 were gradually decreased with the increase of WWP2 expression. Mechanistically, our results indicated that WWP2 interacted with CXCR4 and degraded CXCR4 via the deubiquitination pathway using Co-IP and ubiquitination experiments. There had been reported that CXCR4 inhibition protected cartilage in OA animal models and considered a novel target for OA drug development. 35 Inhibition of CXCR4 and stromal cell-derived factor-1 (SDF-1)/CXCR4-induced chondrocyte autophagy could mitigated OA development.36,37 CXCR4 upregulation overturned the protective effect of miR-373-3p overexpression on IL-1β-induced chondrocyte injury, 38 which supported our finding that CXCR4 overexpression further aggravated FSS-induced cell apoptosis, extracellular matrix degradation and inflammation. Moreover, inhibition of SDF-1α/CXCR4 signaling axis could restrain cartilage degeneration in post-traumatic osteoarthritis rats, 39 which supported our results that CXCR4 was involved in abnormal force-induced OA.
In conclusion, our findings demonstrated that overexpression of PHF8 evidently rescued abnormal force-induced chondrocyte damage. Mechanistically, PHF8, an epigenetic modifier, regulated mechanical force-related OA via modulating the demethylation of H3K9me2 of WWP2 promoter. These results provide an underlying target for OA therapy through interfering with the PHF8-WWP2-CXCR4 signaling pathway.
Supplemental Material
Supplemental Material - Histone demethylase PHF8 protected against chondrocyte injury and alleviated posttraumatic osteoarthritis by epigenetically enhancing WWP2 expression
Supplemental Material for Histone demethylase PHF8 protected against chondrocyte injury and alleviated posttraumatic osteoarthritis by epigenetically enhancing WWP2 expression by Xin Tang, Jingsheng He, and Ye Hao in Human & Experimental Toxicology.
Footnotes
Authors’ contributions
Xin Tang performed the experiments and prepared the manuscript; Jingsheng He analyzed the data and helped improving the manuscript; Ye Hao participated in the study design and conceptualization, supervised and directed the work, drafted and revised the manuscript. All authors have reviewed and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the Natural Science Foundation of Shaanxi Province (2021JM-234).
Ethical statement
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.