
Abstract
Angiotensin II (AngII) is involved in the pathogenesis of hypertensive artery remodeling by inducing a phenotypic switch in vascular smooth muscle cells [Gly14]-Humanin (HNG), a humanin analogue, exerts potent cytoprotective effects both in vitro and in vivo. This study aimed to investigate the effects of HNG on an AngII-induced phenotypic switch in VSMCs and the potential mechanisms underlying these effects. The roles of [Gly14]-Humanin in AngII-stimulated VSMCs proliferation and migration was detected by CCK-8 assay, Cell cycle analysis, wound healing assay, trsnswell assay and western blot. The mechanism by which [Gly14]-Humanin regulates VSMC phenotypic switch was determined by intracellular oxidative stress detection, transcriptomic analysis and qRT-PCR. The results showed that HNG inhibited AngII-induced VSMC proliferation and migration and maintained a stable VSMC contractile phenotype. In addition, HNG reduced the level of AngII-induced oxidative stress in vascular smooth muscle cells. This process could be accomplished by inhibiting nicotinamide adenine dinucleotide phosphate oxidase activity. In conclusion, the results suggested that HNG ameliorated intracellular oxidative stress by inhibiting NAD(P)H oxidase activity, thereby suppressing the AngII-induced VSMC phenotype switch. Thus, HNG is a potential drug to ameliorate artery remodeling in hypertension.
Introduction
Hypertension is the most important risk factor for the development of cardiovascular diseases. 1 Deaths from cardiovascular diseases caused by hypertension have taken first place among the causes of death in China. Artery remodeling is both an adaptive pathological change throughout hypertension and a major cause of irreversible progression of hypertension and its target organ damage in the heart, brain, and kidneys.2,3 It includes early endothelial dysfunction and abnormal proliferation, migration, and apoptosis of smooth muscle cells. It also involves irregular activation, proliferation, transformation, and apoptosis of outer membrane fibroblasts. The entire remodeling process is accompanied by massive extracellular matrix synthesis, degradation, and rearrangement.4–6 However, the molecular mechanisms by which artery remodeling occurs in hypertension have not been fully elucidated yet.
The functional changes in smooth muscle layer of vessel during artery remodeling are currently of wide interest. Existing studies suggest that differentiated mature vascular smooth muscle cells (VSMCs) exhibit extremely low proliferation and migration rates, but they have strong phenotypic plasticity and undergo abnormal phenotypic switch upon stimulation, thus playing an important role in the development of artery remodeling.7–10 Early changes in mechanical stress in the vessel wall, angiotensin II (Ang II), and growth factors stimulate VSMCs to proliferate and migrate phenotypically.11–14 Then, with the involvement of large inflammatory cell infiltration within the vessel wall, excessive inflammatory factors, and adhesion molecules, VSMCs switch to an apoptotic phenotype.15,16 The apoptotic VSMCs further increase inflammatory accumulation within the vessel wall. 17 In summary, artery remodeling is a major pathological alteration in hypertension, and VSMC phenotypic switch is a key process of artery remodeling. The inhibition of VSMC phenotypic switch may be a potentially important therapy to improve artery remodeling and hypertension.
Current studies have found that some of the drugs used in clinical practice can ameliorate artery remodeling, including drugs targeting the renin–angiotensin–aldosterone system (RAAS), calcium channel blockers, statins, aspirin, and so forth.18–22 In addition, some cytokines, such as interleukin-10 (IL-10), endothelial diastolic factor nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF), prostaglandin I2 (PGI2), active substance choline, antioxidant vitamin E, and a series of microRNAs are involved in regulating VSMC phenotypic switch and thus inhibiting artery remodeling.23,24
However, the treatments currently used in clinical practice cannot completely block artery remodeling. Artery remodeling is an ongoing progressive process with multifactorial effects. Moreover, the drugs used clinically are not primarily aimed at inhibiting artery remodeling. The conventional doses of these drugs are much smaller than the doses that can completely inhibit artery remodeling in tissues. Therefore, we needed to explore new ideas and methods for targeted intervention in the artery remodeling process.
Research on endogenous peptides has received much attention in recent years. Endogenous peptides are important regulators of the vital activities of the body. Glucagon-like peptide-1 inhibits VSMC dedifferentiation by regulating mitochondrial dynamics, thereby inhibiting artery remodeling. 25 Angiotensin 2 inhibits VSMC proliferation and adhesion. 26 Osteomorphic protein–peptide inhibited VSMC migration by regulating microRNA expression. 27 Therefore, we further investigated the pathways regulating VSMC phenotypic switch from the perspective of endogenous peptides.
Mitochondria-derived peptides are a series of endogenously secreted peptides encoded by mitochondrial DNA, with humanin being the first type identified.28,29 Humanin consists of 24 amino acids, and glycine (Gly) replaces serine (Ser14) at position 14 of the humanin peptide chain to form [Gly14]-humanin (HNG), which has 1000-fold greater cytoprotective activity than humanin. 30 Humanin is widely distributed in various tissues such as heart, blood vessel wall, kidney, skeletal muscle, and colon. 31 It exhibits anti-oxidative stress, anti-apoptotic, and anti-inflammatory effects in vivo and in vitro32–35 Humanin has recently been found to have an important role in the development of cardiovascular diseases. It can delay the progression of atherosclerosis in several ways, including exerting anti-oxidative stress,36,37 anti-endothelial cell apoptosis,36,38 and anti-inflammatory response effects39–42 and reducing Oxidized Low Den-Sity Lipoprotein (Ox-LDL) accumulation. 43 A series of studies have shown that humanin alleviates myocardial ischemia–reperfusion injury via anti-oxidative stress effect and maintenance of mitochondrial functional homeostasis.44–48 Humanin was shown to inhibit myocardial fibrosis by downregulating myocardial pro-fibrotic cytokine expression to suppress myocardial interstitial fibrosis and fibroblast proliferation.49,50 In addition, humanin prevented intrarenal microartery remodeling in hypercholesterolemic apolipoprotein E (ApoE)-deficient mice. 51 A review of the literature revealed that plasma humanin levels were significantly lower in the elderly group than in the younger group in the healthy population. 52 The humanin levels in circulating blood were significantly lower in patients with microvascular endothelial cell dysfunction. 52 In vitro studies also found reduced humanin gene expression in human umbilical vein endothelial cells (HUVECs) stimulated with high concentrations of Ox-LDL. 35 However, the effect of humanin on VSMC function in hypertensive disease models is still unknown.
The purpose of this study was to investigate the possibility that humanin regulated VSMC phenotypic switch to inhibit artery remodeling. It showed that humanin inhibited the proliferation, migration, and expression of inflammatory factors in AngII-stimulated VSMCs. The mechanism might be related to ameliorate intracellular oxidative stress by regulating the activity of nicotinamide adenine dinucleotide phosphate oxidase [NAD(P)H] oxidase (NOX), thereby inhibiting VSMC phenotypic switching.
Materials and methods
Reagents and antibodies
The following reagents and antibodies were used in the study: (Gly14)-Humanin (absin, #abs45131217), angiotensin II (MCE, #HY-13948), LPS 0127:B8 (Sigma–Aldrich, #L3129), β-actin–HRP (1:5000, Abways Technology, AB2001), anti-alpha smooth muscle actin antibody (1:1000, Abcam, #ab7817), anti-TAGLN/transgelin antibody (1:1000, Abcam, #ab14106), anti-calponin 1 antibody (1:1000, Abcam, #ab46794), anti-MMP2 antibody (1:1000, Abcam, #ab92536), anti-MMP9 antibody (1:1000, Abcam, #ab76003), anti-CDK2 antibody (1:1000, Abcam, #ab32147), anti-CDK4 antibody (1:500, Abcam, #ab199728), anti-cyclin D1 antibody (1:1000, Abcam, #ab16663), anti-cyclin E1 antibody (1:1000, Abcam, #ab71535), anti-rabbit IgG-HRP (1:2000, CST, #70742), and anti-mouse IgG-HRP (1:2000, CST, #7076).
Culture of smooth muscle cells
Rat aortic smooth muscle cells were purchased from ScienCell (#R6110, Shanghai, China). VSMCs were cultured in a smooth muscle cell culture medium containing basal culture medium, 2% fetal bovine serum (FBS, Gibco), 1% smooth muscle cell growth supplement, and 1% penicillin/streptomycin solution (P/S). VSMCs were cultured in a cell culture incubator at 37°C in the presence of 5% CO2, and in an environment of 95% humidity. VSMCs cultured in the second to fifth generations were used in the experiment. The culture medium was changed every 2–3 days. When the cell density was 90%–95%, the cells were digested, centrifuged using 0.25% trypsin, and then passaged. Depending on the grouping, the cells were pretreated with HNG dissolved in SMCM at different concentrations for 12 h. AngII was subsequently dissolved in basal medium containing 0.05% FBS and treated for 24, 48 or 72 h.
Cell viability assay
A CCK-8 cell counting kit (#C0037, Beyotime Biotechnology, China) was used to determine the proliferation viability of VSMCs. Then, 200 μL of cell suspension was added to each culture well of a 96-well plate with 5000 cells per well. Five subwells were made for each treatment, and phosphate-buffered saline (PBS) was rinsed three times at the end of the treatment time. Next, 10 μL of CCK-8 solution was added to each well for 2 h. The optical density at 450 nm was measured and recorded.
Cell cycle analysis
The cells were washed once with pre-cooled PBS. Next, 1 mL of pre-cooled 70% ethanol was added to the collected cell precipitate, gently blown and mixed, and placed at 4°C for overnight fixation. After the cell fixation was completed, the cells were washed once again with pre-chilled PBS. An appropriate amount of propidium iodide (PI) staining solution was prepared based on the sample quantity following the instructions of the cell cycle assay kit (#C1052, Beyotime Biotechnology, China). Then, 0.5 mL of PI staining solution was added to each tube of cell samples. The cell precipitate was resuspended slowly and incubated at 37°C for 30 min in the dark. The precipitate was filtered and assayed using flow cytometry to detect red fluorescence at an excitation wavelength of 488 nm. The cell cycle analysis was performed using FlowJo software.
Wound healing assay
VSMCs (6 × 105/well) were added in the six-well plate, the smooth muscle cell culture medium was washed off, and the cells were synchronized in Dulbecco’s Modified Eagle Medium (DMEM) with 0.05% FBS for 12 h. A sterile pipette tip was used to scratch the monolayer of cells perpendicular to the marker line to obtain a cell-free zone. The cells were rinsed twice with PBS to wash away the detached cells. The cell culture medium was replaced with a new basal medium containing 0.05% FBS, and different treatment drugs were added separately. The scratches were photographed and recorded using a fluorescence microscope (Nikon, Minato, To’kyo, Japan) after 12 and 24 h. The migration distances were measured using ImageJ software.
Transwell assay
The cells were digested with 0.25% trypsin, resuspended in basal culture medium containing 0.05% FBS, counted, and diluted to 5 × 104 cells/100 μL. The cell suspension was added to the upper chamber of the Transwell plate, and 800 μL of basal culture medium containing 20% FBS was added to the lower chamber of the Transwell. Transwell culture plates were incubated in a cell culture incubator at 37°C in the presence of 5% CO2 and 95% humidity for 12 h. The chambers were fixed with pre-chilled 4% paraformaldehyde for 30 min. The cells were gently wiped off the surface of the upper chamber membrane with a wet cotton swab and rinsed three times with PBS, followed by staining with DAPI for 5 min avoiding light. Again, the surface of the upper chamber membrane was gently wiped off with a wet cotton swab. The stained membranes were placed under a fluorescence microscope (Nikon, Minato, To’kyo, Japan) for observation.
Measurement of cytokines by ELISA
VSMCs (6 × 105/well) were maintained in a six-well plate with basal culture medium containing 0.05% FBS for 12 h. Next, they were treated with HNG, AngII, and HNG + AngII for 48 h. The cell culture medium was collected and centrifuged to remove. The concentrations of TNF-α(#EK382), IL-1β(#EK301 B), IL-6 (#EK306), and IL-10 (#EK310) in the culture medium were detected following the instructions of the corresponding ELISA assay kits (Multi Science, Shanghai,China).
Western blot analysis
After the cell treatment was completed, the cell culture was aspirated and rinsed twice with PBS. An appropriate amount of RIPA lysis solution containing 1% protease inhibitor was added to the cell samples. After sufficient lysis and collection of protein samples, the samples containing equal amounts of total protein (30 μg) were separated using SDS-PAGE with 6%–12.5% resolving gel depending on the molecular weight of the target protein to be detected and transferred to PVDF membranes using the electrotransfer method. The membranes were blocked with 5% skimmed milk for 1 h at room temperature and incubated with primary antibodies overnight at 4°C. After recovering the primary antibodies, the membranes were rinsed three times with TBST for 10 min each and then incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. The membranes were rinsed again in the same way. Finally, the protein bands were exposed and analyzed for grayscale values using a fully automated chemiluminescence imaging system.
Intracellular reactive oxygen species staining
A reactive oxygen species (ROS) assay kit (#S0033 M, Beyotime Biotechnology, China) was used to detect intracellular ROS levels. VSMCs (6 × 105/well) were maintained in six-well plates and treated with different drugs. Then, the ROS assay was performed using the fluorescent probe DCFH-DA. Further, DCFH-DA was diluted with serum-free culture medium at a dilution of 1:1000 to a final concentration of 10 μM. The cell culture medium was then removed, and 1 mL of diluted DCFH-DA was added to each well and incubated at 37°C for 20 min. The cells were washed three times with pre-cooled PBS. Then, six-well plates were placed under a fluorescence microscope (Nikon, Minato, To’kyo, Japan), and the fluorescence intensity was visualized and measured using ImageJ software.
Detection of intracellular MDA and SOD
The intracellular malondialdehyde (MDA) levels were measured using a Lipid Peroxidation MDA Assay Kit (#S0131 M, Beyotime, Shanghai, China) according to the manufacturer’s directions. Briefly, 1 × 106 cells were seeded in a 6-well plate. At the end of treatment, cells were collected, lysed by cell lysis buffer and centrifuged at 10,000 ×g for 15 min. The supernatants were reacted with thiobarbituric acid (TBA), and the reaction products were measured spectrophotometrically at 535 nm. The experiment was repeated three times, and the MDA levels were expressed as μmol/mg protein. The intracellular SOD levels were measured using a Total Superoxide Dismutase Assay Kit with WST-8 (#S0101 M, Beyotime, Shanghai, China) according to the manufacturer’s directions.
qRT-PCR
The total mRNA was extracted from the cells with TRIzol reagent (15596018, Invitrogen, Carlsbad, USA). The cDNA was obtained using revere transcription kit (FSO-301,TOYOBO, Osaka, Japan), amplified using SYBR Premix Ex Taq (RR420 A, Takara, Tokyo, Japan), and the signal was detected by QuantStudio Design and Analysis Software. Primers were searched in the PrimerBank Web site (https://pga.mgh.harvard.edu/primerbank/), and synthesized in the GENWIZ company (Suzhou, China).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.0 Software (GraphPad, CA, USA). The results were expressed as mean ± standard deviation. One-way analysis of variance or a two-tailed Student t test was performed to analyze the statistically significance. p values <0.05 indicated a statistically significant difference (*p < 0.05; **p < 0 01; ***p < 0.001; ****p < 0.0001; #p < 0.05; ##p < 0 01; ###p < 0.001; ####p < 0.0001).
Results
HNG inhibited AngII-stimulated VSMC proliferation and suppressed AngII-induced cell cycle progression
VSMCs were treated with 0.1 μM, 1 μM, and 10 μM HNG for 24 and 48 h. The cell proliferation viability data exhibited that HNG was not significantly cytotoxic to VSMCs for the aforementioned concentration range and treatment times (Figure 1(a)). HNG alleviates AngII induced VSMC proliferation. (a) VSMC were treated with 0.1–10 μM HNG for 24 h and 48h, respectively, and cell viability was measured by CCK8 assay. (b) VSMC were treated with AngII at concentrations of 10-8-10-4 mol/L for 24 h, 48 h, and 72 h, respectively, and cell viability was measured by CCK8 assay. (c–e) VSMC were pretreated with 0.1–10 μM HNG for 12 h, followed by treatment with AngII for 24–72 h, and cell viability was determined by CCK8 assay. The measurement data are here expressed as mean ± standard deviation. *p < 0.05, ***p < 0.001 versus the control group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus the AngII group.
VSMCs were treated with AngII in the concentration gradient range of 10−8–10−4M to explore the optimum concentration of AngII for VSMC proliferation. The CCK-8 results showed that cell proliferation significantly increased with AngII treatment at the concentration of 10−6–10−4M for 48 and 72 h. The cells proliferated the most when stimulated with 10−5M AngII (Figure 1(b)). Therefore, we selected 10−5M AngII to stimulate VSMCs and hence establish an in vitro VSMC proliferation-type model.
Next, we pretreated VSMCs with 0.1 μM, 1 μM, and 10 μM HNG for 12 h, and then with AngII for 24 h, 48 h, and 72 h, respectively. Cell proliferation viability data showed that pretreatment of VSMCs with both 0.1 HNG and 10 HNG was effective in reducing the AngII-induced cell proliferation at 48 h and 72 h (Figure 1(c)–(e)).
Furthermore, we detected whether HNG could inhibit AngII-induced cell cycle progression. We found that AngII caused the number of cells in the G0/G1 phase to decrease from 55.6% to 46.0% and the number of cells in the S phase to increase from 9.27% to 26.6%, indicating that AngII enhanced cell cycle progression and promoted cells to enter the S phase. Then, HNG significantly inhibited AngII-induced cell cycle progression, with a decrease in the number of cells in the S phase and an increase in the percentage of cells in the G0/G1 phase (Figures 2(a) and (b)). HNG inhibits AngII-induced cell cycle progression. (a) VSMC were treated with 0.1 μM HNG, 10 μM HNG, AngII, 0.1 μM HNG+Angll, and 10 μM HNG+Angll, then the distribution of cell cycles in each group were detected by flow cytometry. (b) Histogram showed the percentage of cell cycle in each group. Statistical analysis of the differences in S periods for each group. The measurement data are here expressed as mean ± standard deviation. ****p < 0.0001 versus the control group, ####p < 0.0001 versus the AngII group.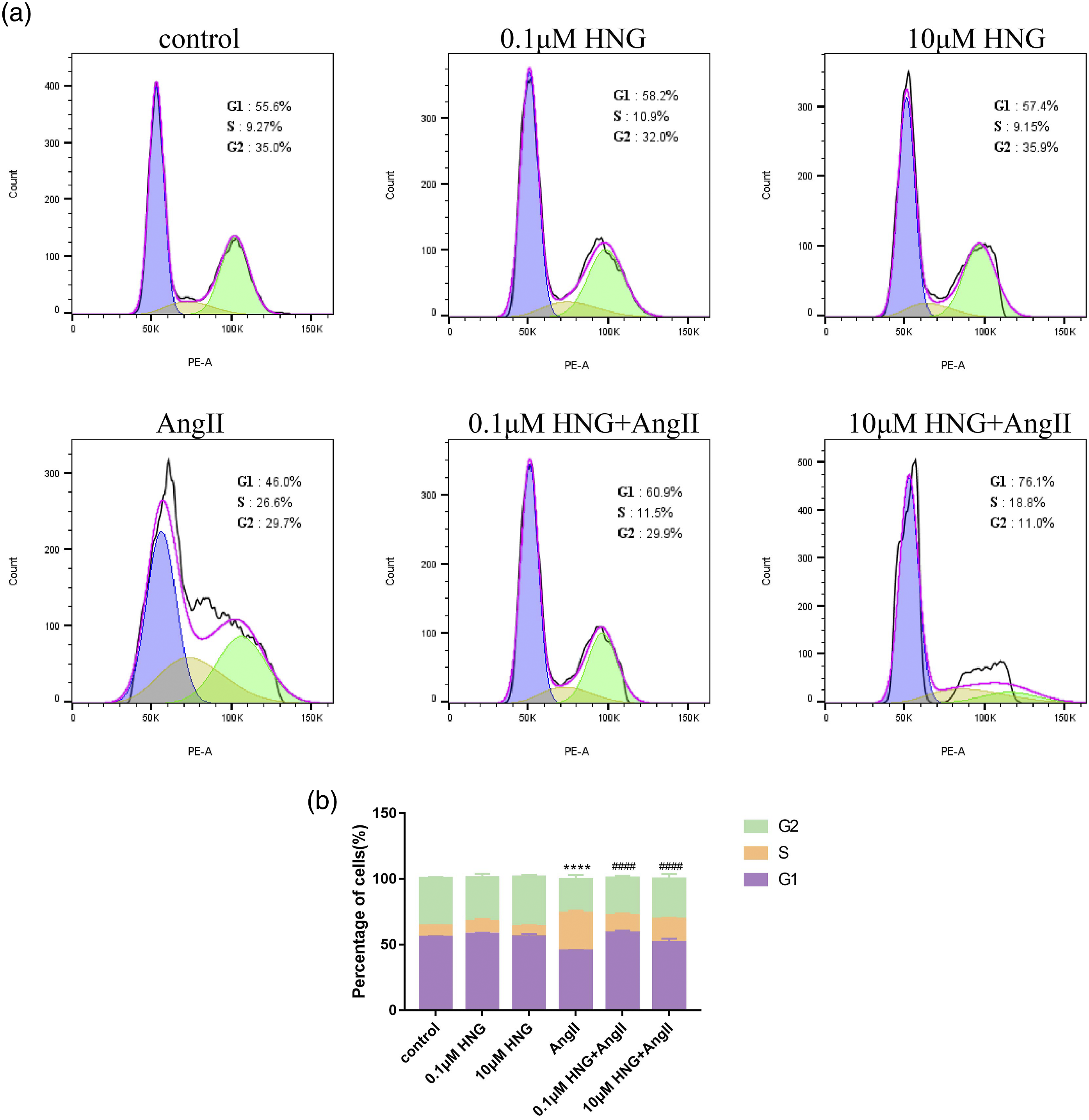
HNG inhibited AngII-stimulated VSMC migration
Another key step in artery remodeling is VSMC abnormal migration. Therefore, we investigated whether HNG could affect VSMC migration. The migration of VSMCs was measured using the wound healing assay. The results showed that AngII significantly increased VSMC migration, while HNG significantly inhibited AngII-induced VSMC migration (Figures 3(a) and (b)). Then, VSMC migration was also assessed by Transwell assay, and the results were consistent with those of the wound healing assay (Figures 3(c) and (d)). Overall, it showed that HNG could effectively inhibit AngII-induced VSMC migration. HNG has an inhibitory effect on AngII-induced VSMC migration. VSMC were co-treated with 0.1 μM HNG and AngII for 24 h, and VSMC migration was measured by wound healing assay (a, b) and Transwell assay (c, d). The measurement data are here expressed as mean ± standard deviation. *p < 0.05, **p < 0.01.
HNG regulated the expression of migration-associated proteins and phenotype transition−associated proteins in VSMC under AngII stimulation
The expression of migration-associated proteins MMP2 and MMP9 in VSMCs was detected by Western blot analysis. The results showed that Angll significantly increased the expression of migration-related proteins MMP2 and MMP9 in VSMCs, and HNG significantly reversed AngII-induced expression of MMP2 and MMP9 compared with AngII alone (Figures 4(c) and (d)). The results of VSMC proliferation−related proteins CDK2, CDK4, cyclinD1, and cyclinE1 showed that AngII significantly increased the expression of CDK4 and cyclinE1, and HNG significantly reversed the AngII-induced expression of CDK4 and cyclinE1 (Figures 4(e) and (f)). HNG reverses AngII regulated the expression of VSMC contractile phenotype-related proteins, and proliferation and migration-related proteins in VSMC. (a, b) The expression of VSMC phenotype transition-related proteins α-SMA, SM22α, and calponin in VSMC after treatment with 0.1μM HNG and AngII for 48 h. (c, d) Expression changes of cell migration-related proteins MMP2 and MMP9 in VSMC after treatment with 0.1 μM HNG and AngII for 48 h. (e, f) Expression changes of CKD2, CKD4, cyclinD1, cyclinE1 in VSMC after treatment with 0.1 μM HNG and AngII for 48 h. The measurement data are here expressed as mean ± standard deviation. ****p < 0.0001 versus the control group, ####p < 0.0001 versus the AngII group.
The shift from the original contractile function to the abnormal behavior of proliferation and migration of VSMCs under AngII stimulation was closely related to the phenotypic switch of VSMCs, which was accompanied by a decrease in the levels of contractile markers. The results showed that the expression of VSMC contractile markers α-SMA, SM22α, and calponin significantly decreased under AngII stimulation compared with that in the control group, while HNG significantly increased the expression of the aforementioned contractile marker proteins in AngII-treated VSMCs (Figures 4(a) and (b)). Therefore, HNG could effectively inhibit AngII-induced VSMC phenotype conversion.
HNG affected the secretion of inflammatory factors in VSMCs under AngII stimulation
AngII has been reported to stimulate VSMCs to secrete a variety of pro-inflammatory cytokines such as IL-6, IL-1β, and TNFα. AngII significantly increased the secretion of IL-6, IL-1β, and TNFα, whereas the secretion of IL-10 was significantly reduced in VSMCs. HNG significantly reversed the AngII-mediated secretion of IL-6, IL-1β, and TNFα (Figures 5(a)–(c)), and the secretion of IL-10 significantly increased in VSMCs (Figure 5(d)). Effect of HNG on the levels of inflammatory factors in VSMC after AngII stimulation. VSMC were treated with 0.1 μM HNG, AngII, 0.1 μM HNG + AngII for 24 h, respectively, and the contents of different inflammatory factors in cell culture supernatants were determined by ELISA. The expression of TNF-α (a); IL-6 (b); IL-1β (c); and IL-10 (d) were measured. The measurement data are here expressed as mean ± standard deviation. *p < 0.05 versus the control group, #p < 0.05 versus the AngII group.
Interleukin-10 (IL-10) is known to have potent, broad downregulatory effects on expression of pro-inflammatory cytokines in various immune and vascular cells 53 and plays an important role in immune homeostasis. 54 A series of studies have shown that IL-10 inhibits TNF-α -induced proliferation of vascular smooth muscle cells.55–57 The concentration of the anti-inflammatory factor interleukin 10 was significantly increased and the pro-inflammatory factor tumor necrosis factor was significantly decreased in the cultures of VSMC pretreated with HNG, showing the significant anti-inflammatory effect of HNG.
HNG ameliorates AngII-induced intracellular oxidative stress
The ROS assay was performed using the fluorescent probe DCFH-DA in the control group, 0.1 μM HNG group, AngII group, and 0.1 μM HNG + AngII group to investigate the effect of HNG on oxidative stress in VSMCs stimulated with AngII. The results showed that AngII significantly induced the production of ROS in VSMCs, while the intracellular ROS levels were significantly inhibited in the HNG and AngII co-treatment groups (Figures 6(a) and (c)). It suggested that HNG could effectively inhibit the AngII-induced increase in the intracellular ROS level in VSMCs and played a protective role against intracellular oxidative stress. HNG ameliorates AngII-induced intracellular oxidative stress response. (a, b) The intracellular ROS levels were measured in VSMC after treatment with 0.1μM HNG and AngII for 48 h. The Rosup was used as a positive control; ROS production was detected by DCFDA-H2 staining. (c) Cellular MDA levels were measured using the TBA method, and the concentrations of MDA were expressed as μmol/mg protein. (d) Assay of SOD activity using a Total Superoxide Dismutase Assay Kit with WST-8.
The results of assaying the concentration of lipid peroxide (malondialdehyde) MDA in different treatment groups showed that AngII induced an increase in intracellular MDA level and a decrease in intracellular MDA level after HNG pretreatment. For the antioxidant SOD activity, the results showed a decrease in SOD activity in the AngII treated group and a significant increase in SOD activity in the HNG pretreated group.
A review of the relevant literature revealed that increased intracellular oxidative stress significantly induced phenotypic transformation of vascular smooth muscle cells. 13 Therefore, we suggest that HNG may inhibit vascular smooth muscle cell phenotype conversion by attenuating intracellular oxidative stress.
RNA-seq analysis revealed different gene expression profiles in HNG-treated VSMCs
RNA-seq was performed in the AngII group, the 0.1 μM HNG + AngII group, and the control group to determine the molecular changes in HNG-treated VSMCs. The heat map results showed that VSMCs in the AngII group versus the control group and 0.1 μM HNG + AngII group versus AngII group showed significantly different gene expression profiles (Figures 7(a) and (b)). The volcano plot showed that 8349 genes (upregulated: 3831; downregulated: 4518) were differentially expressed between the AngII and control groups. In addition, 6884 genes (upregulated: 3779; downregulated: 3085) were differentially expressed between the 0.1 μM HNG + AngII group and the AngII group (Figures 7(c) and (d)). Transcriptomics analysis of HNG and AngII-stimulated VSMC. (a, b) Heatmap showing the differentially expressed genes (DEGs) between the AngII, 0.1 μM HNG+AngII, and control groups. Each column represents a sample and each row represents a gene. (c, d) Volcano plots display up and down regulated mRNA transcripts between AngII versus control groups, and HNG+AngII versus AngII groups. (e, f) Significant enriched Gene Ontology (GO) terms in Ang II versus control groups and of HNG+AngII versus Ang II groups. (g, h) KEGG pathway enrichment analysis of differentially expressed transcripts in the two comparisons of Ang II versus control and of HNG+AngII versus Ang II.
Gene Ontology (GO) databases are used to obtain functional information about these populations and process differentially expressed genes (DEGs) according to their important functions. DEGs are grouped into three main GO functional categories: biological processes, cellular components, and molecular functions. Biological processes, such as “angiogenesis,” “cell proliferation,” and “extracellular matrix organization,” were significantly enriched in the AngII and control groups. In the 0.1 μM HNG + AngII group and AngII group, “inflammatory response,” “regulation of apoptotic processes,” “response to tumor necrosis factor,” “response to tumor necrosis factor,” and the biological processes such as “cellular response to transforming growth factor” were clearly enriched.
KEGG pathway enrichment analysis further revealed the key pathways associated with the identified DEGs. Figure 7(g) and (h). shows the top 30 most enriched pathways in the AngII group versus the control group and the 0.1 μM HNG + AngII group versus the AngII group. We selected the AGE-RAGE signaling pathway in diabetic complications among the differentially expressed signaling pathways enriched between the AngII and control groups and between the HNG+AngII and AngII groups. Among the differentially expressed genes enriched to this signaling pathway were selected those associated with reactive oxygen species production: the NAD(P)H oxidase family proteins NOX1, NOX4. NADPH oxidase was used as the entry point for further mechanism analysis. Effect of HNG on the transcriptional levels of NAD(P)H oxidase and its upstream and downstream signaling molecules in VSMC after AngII stimulation. VSMC were treated with 0.1 μM HNG, AngII and 0.1 μM HNG + AngII for 48 h, respectively. qRT-PCR was used to detect the transcript levels of different signaling molecules. (a) mRNA expression of NAH(P)H oxidases NOX1, NOX2, and NOX4; (b) JAK2, STAT; (c) Migration-related genes MMP2 and MMP9; (d) Proliferation-related genes P53 and CDK4 were measured. The measurement data are here expressed as mean ± standard deviation. *p < 0.05, ****p < 0.0001 versus the control group, #p < 0.05, ####p < 0.0001 versus the AngII group. Schematic diagram showing the role of HNG in AngII-mediated signaling pathway of VSMCs.

HNG inhibited AngII-induced expression of NAD(P)H oxidase
Summary of qRT-PCR primers.

Discussion
Artery remodeling is a major pathological change during hypertension, and VSMC phenotypic conversion is a key process of artery remodeling. The original contractile phenotype is lost and transformed into functions such as proliferation, migration, and extracellular matrix secretion. Humanin is the first mitochondria-derived peptide to be discovered; it has been reported to play cytoprotective roles in leukocytes, germ cells, neurons, and many other cells. 31 The present study was novel in demonstrating that humanin inhibited AngII-induced VSMC migration and cell cycle progression, suppressed VSMC inflammatory secretion, and maintained a stable VSMC contractile phenotype. The present study supported the potential role of humanin in inhibiting hypertensive artery remodeling.
VSMC phenotypic conversion is closely related to oxidative stress. Oxidative stress and mitochondrial dysfunction are is considered to be one of the important causes of VSMC phenotypic transition.13,58,59 Numerous studies have shown that factors such as angiotensin (AngII), inflammatory factors, and mechanical stress on the vessel wall activate intracellular NAD(P)H oxidase in the vessel wall, leading to massive ROS production. Reactive oxygen causes oxidation of NADH, NADPH, and glutathione bound to the mitochondrial bilayer membrane permeability pore, prompting the opening of the mitochondrial bilayer membrane permeability pore. Sustained mitochondrial permeability transition pore opening leads to Ca2+ overload, oxidized mitochondrial glutathione, and elevated levels of reactive oxygen species in mitochondria, resulting in cytochrome C release and decreased and absent mitochondrial membrane potential. This eventually causes apoptosis. Oxidative stress stimulates cis-regulatory elements involved in regulating VSMC phenotypic transition60,61; induces the expression of VSMC phenotypic transition–related transcription factors62,63; affects signaling pathways involved in vascular proliferation, migration, apoptosis, secretion, and inflammation13,64; and ultimately causes hypertensive vascular injury and remodeling. Humanin has a potent protective role in regulating mitochondrial function in many biological processes such as apoptosis, 65 substrate metabolism, 32 inflammatory responses, and oxidative stress.45,66,67 In this study, we performed the transcriptomic analysis of VSMCs in the 0.1 μM HNG + AngII, AngII, and control groups and found that the members of NAD(P)H oxidase family, such as NOX1, NOX2, and NOX4, were significantly differentially expressed in different treatment groups.
We demonstrated using q-PCR that HNG could inhibit AngII-induced NOX expression. Therefore, we speculated that HNG might inhibit ROS production and oxidative stress response by suppressing NAD(P)H oxidase activity.
In addition, we revealed that HNG could repress the expression of JAK2/STAT1. Recent studies have reported that JAK/STAT is a key regulator of NOX1 and NOX4 in VSMCs. The inhibition of the JAK/STAT pathway can suppress Nox-dependent oxidative stress responses. 68 Moreover, the role of humanin in regulating the JAK/STAT signaling pathway to inhibit oxidative stress has been demonstrated in a variety of cells.65,69,70 Therefore, we found that humanin could inhibit intracellular NADPH oxidase activity and suppress ROS production by inhibiting the JAK/STAT signaling pathway. It is one of the possible mechanisms for inhibiting VSMC phenotypic transition.
The present study had several limitations. We showed that one of the mechanisms about HNG might be related to the inhibition of NADPH oxidase activity. However, other mechanisms may be involved in the protective effect of HNG on VSMCs. Second, our study demonstrated the effect of HN in vitro. The inhibitory role of HNG in artery remodeling needs to be validated in vivo.
Conclusion
Our study was novel in demonstrating the role of HNG in the phenotypic regulation of VSMCs. We also demonstrated that HNG might attenuate VSMC intracellular oxidative stress by inhibiting NADPH oxidase activity. The findings might provide new ideas to reduce artery remodeling in hypertension.
Footnotes
Author contributions
L. Jiang, and M. Zhang, designed the project and directed the research. Y. Xie and J.Zhang carried out experiments and wrote the manuscript. M.Zhang obtained and analyzed data. The views and opinions expressed within this manuscript are those of all authors. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (82170450).