
Abstract
Increasing studies indicate that cholesterol plays an important role in drug resistance. ARL4C is implicated in the export and import of cholesterol, therefore this study aimed to explore the effect of ARL4C on the resistance of ovarian cancer (OVC) to Carboplatin. This study collected OVC tissue samples from patients who are sensitive or resistant to carboplatin, and established Carboplatin-resistant OVC cell lines, OVCAR3(R) and SKOV3(R) using OVCAR3 and SKOV3. High throughput sequencing was conducted to find genes that regulated by ARL4C. Cholesterol esterification was performed to evaluate the transport of cholesterol from Lysosome (LY) to Endoplasmic reticulum (ER). The fluorescence of LC3-GFP-mRFP was used to evaluate the function of autophagy flux. As indicated by PCR, western blot and Immunohistochemistry, ARL4C was increased in the Carboplatin-resistant OVC tissues and cells. Knockdown of ARL4C attenuated the resistance of OVCAR3(R) and SKOV3(R) to Carboplatin. By suppressing Notch signal, ARL4C knockdown inhibited the transcriptional function of RBP-Jκ and RBP-Jκ-induced H3K4Me3, which collectively reduced OSBPL5 expression. OSBPL5 deficiency inhibited the transport of cholesterol from LYs to ER, which led to the accumulation of cholesterol in LYs and the dysfunction of autophagy. In summary, ARL4C knockdown attenuated the resistance of OVC to Carboplatin by disrupting cholesterol transport and autophagy. This study revealed a promising target to attenuate the resistance of OVC to Carboplatin and elucidated the potential mechanism.
Introduction
Ovarian cancer (OVC) is the third most common cancer in women, and its mortality ranks first among gynecological malignancies. 1 Due to the lack of specific symptom in the early stage of OVC, 70% of patients are in the advanced stage when they have been diagnosed with OVC. Platinum drugs, such as Cisplatin and Carboplatin, are commonly used in the first-line chemotherapy of OVC. The cytotoxic mechanism of platinum drugs is associated to the DNA damage, the dysfunction of mitochondria and the activation of apoptotic signal, which lead to cell death. 2 However, most patients finally show drug resistance and have poor treatment effect. 3 Therefore, the research on the mechanism of chemotherapy resistance is of great clinical significance to improve the prognosis and survival rate of patients with OVC.
Cholesterol, a derivative of cyclopentane and phenanthrene, widely exists in various cells. It is not only an important component of cell membrane and plasma lipoprotein, but also a precursor compound of many special bioactive molecules, including cholic acid, vitamin D and steroid hormones. However, accumulating evidence indicates that cholesterol is an important inducer for drug resistance.4–8 Elevated cholesterol in mitochondria can block apoptotic signals induced by anticancer drugs. 5 The change of cholesterol content in cell membrane affect the permeability and absorption of chemotherapeutic drugs, through regulating lipid rafts and ABC transporters, such as ABCC1, P glycoprotein and BCRP. 6 Moreover, multiple signals have been found to be regulated by cholesterol.7,8 Among these signals, EGFR-mediated pathways lead to epithelial mesenchymal transformation that promotes stem cell like properties of cancer, thereby enhancing drug resistance. 7
ADP ribosylation like factor 4C (ARL4C), also known as ARL7, is one of the subfamily members of ADP ribosylation factor (ARF). ARL4C plays an important role in vesicle transport, which is critical for ABCA1-mediated cholesterol secretion pathway. 9 High expression of ARL4C has been confirmed as an independent predictive factor for worse prognosis in endometriosis-associated OVC. 10 Moreover, ARL4C is associated with initiation and progression of lung adenocarcinoma and gastric cancer.11,12 Increased expression of ARL4C enhances the resistance of pancreatic cancer to Gemcitabine. 13 All these data suggested cancer-promoting effects of ARL4C.
Endocytosis is a primary pathway for the uptake of cholesterol. 14 Endocytic cholesterol initially enters into late endosomes/lysosomes (LEs/LYs) for the hydrolysis of cholesteryl esters to release free cholesterol. The released free cholesterol from LEs/LYs is transported to endoplasmic reticulum (ER), plasma membrane and mitochondrion for the regulatory, structural and metabolic functions. OSBPL5 (also name as ORP5) belongs to a member of the oxysterol-binding protein (OSBP) family. OSBPL5 cooperates with NPC1 to release cholesterol from LEs/LYs. 15 OSBPL5 depletion leads to the retention of cholesterol in LEs/LYs. 15 The cholesterol retention in LEs/LYs not only suppresses the biological functions of cholesterol in cells, but also impairs the fusion of autophagosomes (APs) with LYs to form autolysosomes, resulting in a defective autophagic flux. In our preliminary experiment, OSBPL5 expression was down-regulated in OVC after knockdown of ARL4C. This result implied that ARL4C is also involved in intracellular cholesterol transport, in addition to the export and secretion of cholesterol.
Based on previous studies, we aimed to determine the effect of ARL4C on the sensitivity of OVC cells to Carboplatin in this study. Since ARL4C is closely linked to cholesterol transport, it was conjectured that the effect of ARL4C on OVC cells is associated to the regulation of cholesterol. This hypothesis was identified in this study.
Materials and methods
Tissue collection and immunocytochemistry analysis
This study collected OVC tissue samples from three patients who are sensitive to Carboplatin. The tumor volume was notably shrunk after treatment with Carboplatin. The OVC tissues were obtained by surgical resection in the early stage of the chemotherapy with Carboplatin. In addition, we collected OVC tissue samples from five patients who are resistant to Carboplatin. Carboplatin fails to effectively suppress the growth of OVC in the late stage of chemotherapy. The research protocol was approved by the Ethics Committee of Hunan Provincial Tumor Hospital (Changsha, China), and all patients involved provided written informed consent.
OVC tissues were initially fixed in 10% formalin. Paraffin-embedded sections from the tissue specimens were deparaffinized and heated at 97°C in 10 mM citrate buffer (pH 6.0) for 20 min to retrieve antigens. Each tissue sample was cut to eight sections for immunocytochemistry analysis. Tissue sections were incubated with anti-ARL4C antibody (1:200; Abcam, Cambridge, MA, USA) overnight at 4°C followed by horseradish peroxidase (HRP)-labelled anti-rabbit IgG (1:500; Abcam) at room temperature for 30 min. Each section was immersed a diaminobenzidine working solution at room temperature for 3–10 min.
Generation of carboplatin-resistant OVC cell lines
OVCAR3 and SKOV3 cells (American Type Culture Collection; Manassas, VA, US) were seeded into 10-cm dishes and cultured to more than 85% confluence. Carboplatin was added to the culture medium to the concentration of 12 μM. Carboplatin induced massive cell death, but there were a few of cells that were surviving. The survived cells were isolated and expanded in fresh culture medium without Carboplatin until to 85% confluence. The confluent cells were subjected to more rounds of Carboplatin selection. Finally, the Carboplatin tolerant cells were used as the resistant cells, named as OVCAR3(R) and SKOV3(R). The original OVCAR3 and SKOV3 cells were treated as the Carboplatin-sensitive cells, and renamed as OVCAR3(S) and SKOV3(S). Cell viability and apoptosis assays were conducted to evaluate the sensitivity of OVCAR3(S), SKOV3(S), OVCAR3(R) and SKOV3(R) to Carboplatin.
Transfection
shRNAs against ARL4C and RBP-Jκ were obtained by GenePharma Co., Ltd. (Shanghai, China). shRNA-ARL4C and shRNA-RBP-Jκ were transfected into OVCAR3(R) and SKOV3(R) cells using the Lipofectamine 3000 kit (Invitrogen, Shanghai, China) according to the manufacture’s instruction. In addition, ABCA1, OSBPL5 and RBP-Jκ expression vectors were constructed using enhanced green fluorescent protein plasmid-C1 vector (GenePharma). Transfection of expression vectors also used Lipofectamine 3000 kit. Cells were collected 48 h after the transfection.
Cell viability assay
Cells, which underwent indicated transfection, were exposed to different dosages of carboplatin. Cells were washed twice using PBS and further incubated with MTT [3-(4,5-dimethylthia-zolyl-2)-2,5-diphenyltetrazolium bromide] for 2 h. The blue formazan crystal was solubilized with DMSO (100 μL/well), and the absorbance of blue-dyed solution was read at a wavelength of 550 nm.
Apoptosis assay
Cells, which underwent indicated transfection, were exposed to 12 μM carboplatin. Cells were resuspended in phosphate-buffered saline (PBS) and mixed with Annexin V-fluorescein isothiocyanate (FITC)/Propidium Iodide (PI) (Dojindo Molecular Technologies, Inc., Dalian, China). After incubation for 15 min in the dark, apoptosis were analysed using a FACS Calibur Flow Cytometer (Beckman Coulter, CytoFLEX S, United States).
Invasion assay
Cells were seeded in the upper chamber of Transwell plates (Corning, Shanghai, China) to evaluate cell invasion capacity. FBS-containing culture medium was added to the lower chamber. The filters were removed and washed in PBS. Cells, which invaded through the upper chamber lower surface, were fixed in cold methanol for 10 min and stained with haematoxylin. Finally, cells were counted in 10 fields at 200× magnification.
Quantitative polymerase chain reaction
The primer information in PCR assay.

Western blot and immunocoprecipitation (Co-IP) assays
The proteins were separated on 8–12% SDS polyacrylamide gels and then transferred to PVDF membranes (Pierce; Thermo Fisher Scientific, Inc.). The membranes were blocked with 5% skimmed milk and subsequently incubated overnight at 4°C with antibodies targeting ARL4C (1:200; Abcam), ABCA1(1:200; Abcam), OSBPL5 (1:200; Abcam), RBP-Jκ (1:100; Abcam), ULK1 (1:100; Santa Cruz Biotechnology, Santa Cruz, California, USA), LC3 (1:100; ptgcn, Wuhan, China), p62 (1:100; ptgcn), NICD1 (1:200; Santa Cruz Biotechnology), NICD4 (1:100; Santa Cruz Biotechnology), Hey1 (1:100; Abcam), Hes1 (1:100; Abcam), KDM6B (1:200; Abcam), and GAPDH (1:500; ptgcn). Horseradish peroxidase-conjugated secondary antibodies and ECL chemiluminescence (Pierce, Thermo Scientific, Rockford, IL, USA) were added to observed the blots on the membranes.
For Co-IP, cell lysates were incubated with primary antibody against RBP-Jκ at 4°C overnight. Protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology) were used to extract RBP-Jκ-protein complex from cell lysates, according to the manuscript instruction. Thereafter, the beads were isolated by rinsing with lysis buffer, and the enrichment of NICD4 in RBP-Jκ-protein complex was finally analyzed by western blot.
Dual-luciferase reporter assay
OSBPL5 gene promoter fragment was cloned into pGL4 vector (Addgene, Inc., Cambridge, MA, USA) upstream of the firefly luciferase coding region within restriction sites XhoI and NotI (Takara Bio, Inc., Otsu, Japan). Corresponding mutation was introduced into the RBP-Jκ binding site by site directed mutagenesis using a fast mutation kit (New England BioLabs, Inc., Ipswich, MA, USA). The wild type and mutant type of reporters were co-transfected with RBP-Jκ overexpression vector using LipofectamineTM 2000. Cells were lysed 24 h using Glo Lysis Buffer (Promega) after transfection, and the luciferase activity was analyzed using Bright-GloTM Luciferase Assay System (Promega).
Chromatin immunoprecipitation assay
Cells were treated with ChIP lysis buffer (Thermo Fisher Scientific, Inc.) to extract total chromatin. The chromatin was sonicated to an average size of 250–500 bp using an immersion sonicator, and then incubated overnight with the Tri-Methyl-Histone H3 (Lys4) (H3K4Me3) antibody (Santa Cruz Biotechnology) or IgG at 4◦C. Protein A/G sepharose-beads (Thermo Fisher Scientific, Inc.) were used to obtain H3K4Me3-DNA complex, and consequently the DNA fraction in OSBPL5 gene promoter was analysed using qPCR. DNA enrichment was expressed as fold enrichment of specific binding over the control (nonspecific IgG binding).
Analysis of autophagic flux
Cells were transfected with a fluorescent mRFP‐GFP‐tagged LC3 plasmid 24 using Lipofectamine 2000. The expression of GFP and mRFP was visualized with a laser scanning confocal microscope (Olympus, Tokyo, Japan).
Cholesterol measurement
Cellular free cholesterol concentration was measured using a Cholesterol Assay Kit according to manufacturer’s instructions (ab65390, Abcam). In brief, cells were lysed and incubated with cholesterol oxidase that acts on free cholesterol to produce a chemical which reacts with a probe to generate color (570 nm) and fluorescence (Ex/Em = 538/587 nm). The values were normalized to the total cellular protein levels, which were determined with a BCA protein assay kit (Thermo Fisher Scientific).
In addition to free cholesterol, total cholesterol in cells also contains cholesterol esters. For the detection of the total cholesterol, cholesterol esterase is used to hydrolyze cholesteryl ester into free cholesterol. Afterwards, free cholesterol concentration was measured used the method mentioned above. The amount of cholesterol ester can be calculated by subtracting free cholesterol from total cholesterol.
Cholesterol esterification
Cells were starved of cholesterol in culture medium with low level of FBS, and treated with radio-labeled fatty acids ([1–14C]-palmitate) alone or in combination with LDL. After incubation for 6 h, cellular lipids were extracted and the formation of radio-labeled cholesteryl esters was visualized by exposing to a BAS-MS imaging plate (Fujifilm). The relative intensities of bands corresponding to cholesteryl ester were quantified using ImageJ Software (Fujifilm).
Statistical analyses
All experiments were performed at least three times. Experimental data were examined using either a one-way ANOVA or an unpaired two-tailed t-test with GraphPad Prism software (GraphPad Software, San Diego, CA, USA). A value of p < 0.05 was set as the statistical significance level.
Results
The expression of ARL4C is up-regulated in platinum resistant OVC tissues
As indicated by the data in TCGA database, ARL4C is up-regulated in OVC compared to the normal ovarian tissue (Figure 1a and b). Moreover, ARL4C expression is higher in OVC than other types of cancers (Figure 1c). The expression of ARL4C in OVC is higher in OVC with higher stage (Figure 1d). However, it is unclear the expression of ARL4C in OVC sensitive or resistant to Carboplatin. This study collected OVC tissue samples from five patients resistant to Carboplatin and from three patients sensitive to Carboplatin. PCR, western blot and IHC assays were performed to detect the expression of ARL4C in the tissue samples. As indicated by PCR and western blot, ARL4C mRNA and protein levels are much higher in Carboplatin-resistant OVC tissues than in Carboplatin-sensitive OVC tissues (Figure 1e and f). Immunocytochemistry analysis further indicated that ARL4C protein level is highly expressed in the cancer cells rather than the stroma cells in Carboplatin-resistant OVC tissues. Moreover, ARL4C protein is mainly distributed in the cytoplasm of OVC. We performed study in vitro to further investigate the effect of ARL4C on the sensitivity of OVC to platinum. ARL4C is up-regulated in platinum resistant OVC tissues. (a) Data from TCGA show ARL4C expression in various tumors and the corresponding normal tissues; (b) Data from TCGA show ARL4C expression in OVC and the normal ovarian tissue; (c) ARL4C expression in various tumors; (d) The expression of ARL4C in OVC each stage of OVC; This study collected OVC tissue samples from five patients resistant to Carboplatin and from three patients sensitive to Carboplatin. (e) PCR, (f) western blot and (g) IHC assays were performed to detect the expression of ARL4C in the tissue samples. IHC results showed two representative tissue samples of both Carboplatin-sensitive and resistant OVC. ***p < 0.001 vs. OV(S). OV(S): Ovarian cancer sensitive to Carboplatin; OV(R): Ovarian cancer resistant to Carboplatin.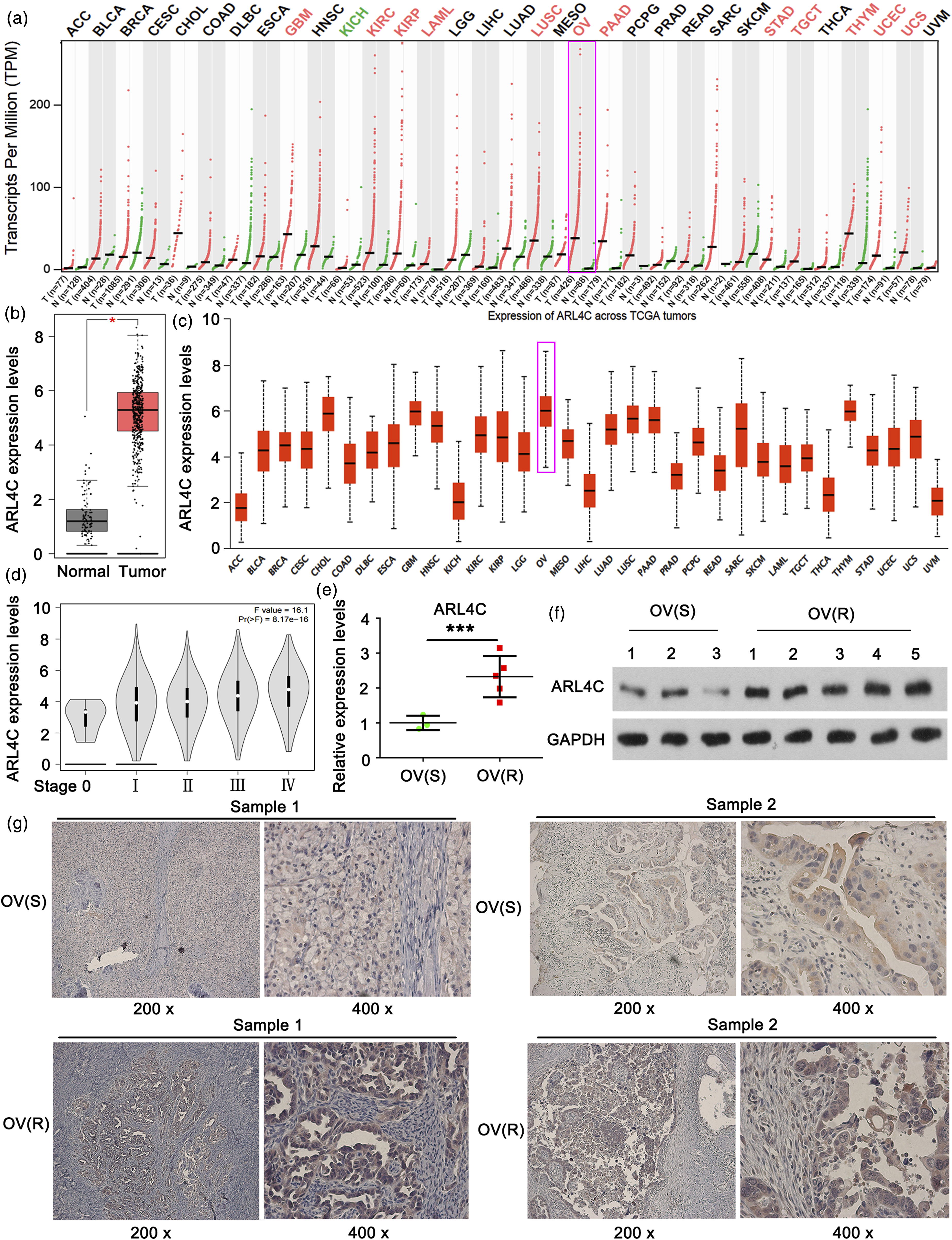
ARL4C knockdown increases the sensitivity of OVC to carboplatin
We established Carboplatin-resistant OVC cells. ARL4C expression was higher in OVCAR3(R) and SKOV3(R) than in OVCAR3(S) and SKOV3(S) (p < 0.001, Figure 2a). We designed three shRNAs for ARL4C knockdown. shRNA3 conferred the most significant effect on the knockdown of ARL4C in OVCAR3(R) and SKOV3(R) among three shRNAs (p < 0.001, Figure 2b). shRNA3 was used in the following study. Western blot analysis confirmed that ARL4C protein levels were higher in OVCAR3(R) and SKOV3(R) than in OVCAR3(S) and SKOV3(S) but knockdown of ARL4C decreased the protein levels in OVCAR3(R) and SKOV3(R) (Figure 2c). In cytotoxicity test, OVCAR3(R) and SKOV3(R) had higher cell viability than OVCAR3(S) and SKOV3(S) when exposing to same dosage of Carboplatin (Figure 2d). Transfection with ARL4C-shRNA3 decreased cell viability of OVCAR3(R) and SKOV3(R) subjected to Carboplatin treatment. The IC50 of Carboplatin in OVCAR3(S) and SKOV3(S) were used in following apoptosis and cell invasion tests. The apoptosis rate of OVCAR3(R) and SKOV3(R) were lower than those of OVCAR3(S) and SKOV3(S) when exposing to 12 μM Carboplatin (p < 0.001, Figure 2e). However, knockdown of ARL4C elevated the apoptosis rate of OVCAR3(R) and SKOV3(R) upon Carboplatin (p < 0.001). OVCAR3(R) and SKOV3(R) showed increased invasive ability compared to OVCAR3(S) and SKOV3(S) when exposing to 12 μM Carboplatin (p < 0.001, Figure 2f). Silencing ARL4C suppressed the invasion of OVCAR3(R) and SKOV3(R) (p < 0.001). These data suggested that ARL4C knockdown increases the sensitivity of OVC to Carboplatin. ARL4C knockdown increases the sensitivity of OVC to Carboplatin. Carboplatin-resistant OVC cell lines, OVCAR3(R) and SKOV3(R) were established using OVCAR3 and SKOV3. The original OVCAR3 and SKOV3 cells were treated as the Carboplatin-sensitive cells, and renamed as OVCAR3(S) and SKOV3(S). (a) PCR analysis evaluated ARL4C expression in OVCAR3(R), SKOV3(R), OVCAR3(S) and SKOV3(S). (b) We designed three shRNAs for ARL4C knockdown. PCR analysis evaluated ARL4C expression; *p < 0.05, **p < 0.01, ***p < 0.001 vs. NC; (c) Western blot was conducted to detect ARL4C protein level; (d) Cell viability, (e) apoptosis rate and (f) cell invasion were evaluated in OVCAR3(R), SKOV3(R), OVCAR3(S) and SKOV3(S) with ARL4C knockdown or not. ***p < 0.001 vs. OVCAR3(S) and SKOV3(S); ###p < 0.001 vs. OVCAR3(R) and SKOV3(R).
ARL4C knockdown increased total cholesterol and free cholesterol in OVC
Total cholesterol in cells contains free cholesterol and cholesterol esters. It has been verified that ARL4C plays an important role in ABCA1-mediated cholesterol secretion. We want to know whether ARL4C knockdown influences the total cholesterol, free cholesterol and cholesterol esters in OVC. As indicated by PCR and western blot analysis, knockdown ARL4C decreased ABCA1 in SKOV3(R) (p < 0.05, Figure 3a and b), but not in OVCAR3(R). Overexpression of ABCA1 notably increased ABCA1 in OVCAR3(R) and SKOV3(R) with ARL4C knockdown (p < 0.001). Free cholesterol and total cholesterol, but not cholesterol esters, were increased in SKOV3(R) after ARL4C knockdown (p < 0.01, Figure 2c). Overexpression of ABCA1 only moderately suppressed the increase of free cholesterol and total cholesterol in OVC with ARL4C knockdown. This suggested that ABCA1 is unable to completely compensate the function of ARL4C in cholesterol after it knockdown. In general, the increase of cholesterol in cancer cells is associated with enhanced drug resistance. It is difficult to understand why the increase of cholesterol in ARL4C-depleted OVC was in company with increased sensitivity to Carboplatin. ARL4C regulates total cholesterol and free cholesterol in OVC. OVCAR3(R) and SKOV3(R) were subjected to treatments with ARL4C knockdown alone or in combination with ABCA1 overexpression. (a) PCR analysis evaluated ABCA1 expression; (b) Western blot was conducted to detect ABCA1 protein level; (c) Total cholesterol, free cholesterol, and cholesterol esters were detected in SKOV3(R) cells. **p < 0.01 vs. control.
ARL4C regulates intracellular cholesterol transport by regulating OSBPL5
High-throughput sequencing was conducted in OVCAR3(R) and SKOV3(R) cells to find genes whose expression was significantly changed after the knockdown of ARL4C by shRNA2 and shRNA3. Intersection analysis showed 131 similar genes that were changed in both OVCAR3(R) and SKOV3(R) cells after ARL4C knockdown (Figure 4a). GO enrichment analysis showed that cholesterol metabolism was notably changed after ARL4C knockdown (Figure 4b). This result is consistent with our prediction. Among the genes implicated to cholesterol metabolism, we found that ARL4C knockdown decreased OSBPL5 expression. This result was further confirmed by PCR and western blot assay (Figure 4c and d). OSBPL5 is located mostly in ER and takes the responsible for the transport cholesterol from LEs/LYs to ER. We want to know whether the reduction of OSBPL5 caused by ARL4C knockdown influences the transport of cholesterol from LEs/LYs to ER. Cholesterol esterification assay is a standard measurement of cholesterol transport to the ER. In ER, Acyl-CoA:cholesterol acyltransferase (ACAT) induces cholesterol esterification using free cholesterol from LEs/LYs and fatty acids. Therefore, in the assay, OVC cells were treated with radio-labeled fatty acids alone or in combination with LDL (containing cholesterol). The uptake of LDL is through LEs/LYs pathway. The disruption of cholesterol transport from LEs/LYs to ER will suppress cholesterol esterification. As expected, ARL4C knockdown reduced the radio-labeled cholesterol esters (p < 0.001, Figure 4e), while overexpression of OSBPL5 restored the level of radio-labeled cholesterol esters in OVC cells. ARL4C regulates intracellular cholesterol transport by regulating OSBPL5. High-throughput sequencing was conducted in OVCAR3(R) and SKOV3(R) cells to find genes significantly changed after the knockdown of ARL4C by shRNA2 and shRNA3. (a) Intersection analysis showed 131 similar genes that were changed in both OVCAR3(R) and SKOV3(R) cells after ARL4C knockdown. (b) The 131 genes were used for GO enrichment analysis. (c) PCR analysis evaluated OSBPL5 expression after the knockdown of ARL4C by shRNA2 and shRNA3. (d) Western blot analysis evaluated OSBPL5 protein levels after the knockdown of ARL4C by shRNA2 and shRNA3. (e) Cholesterol esterification assay was conducted to evaluate cholesterol transport from LY to the ER. **p < 0.01 and **p < 0.01 vs. control.
OSBPL5 overexpression restored the resistance of OVCAR3(R) and SKOV3(R) to carboplatin after ARL4C knockdown
To determine whether OSBPL5 mediates the effect of ARL4C on the resistance of OVCAR3(R) and SKOV3(R) to Carboplatin, we overexpressed OSBPL5 after ARL4C knockdown. As indicated by PCR and western blot assays, overexpression of OSBPL5 reversed the reduction of OSBPL5 caused by ARL4C knockdown (p < 0.001, Figure 5a and b). In addition, OSBPL5 overexpression restored the cell viability and invasion, and suppressed the apoptosis in ARL4C-silenced OVCAR3(R) and SKOV3(R) (p < 0.01, Figure 5c–e). OSBPL5 overexpression restored the resistance of OVCAR3(R) and SKOV3(R) to Carboplatin after ARL4C knockdown. OVCAR3(R) and SKOV3(R) were subjected to treatments with ARL4C knockdown alone or in combination with OSBPL5 overexpression. (a) PCR analysis evaluated OSBPL5 expression. (b) Western blot analysis evaluated OSBPL5 protein levels. (c) Cell viability, (d) apoptosis rate and (e) cell invasion were evaluated in OVCAR3(R), SKOV3(R), OVCAR3(S) and SKOV3(S) with ARL4C knockdown or not. ***p < 0.001 vs. Carboplatin treatment alone; ##p < 0.01 vs. Carboplatin treatment with ARL4C knockdown.
ARL4C depletion impaired the autophagy by suppressing OSBPL5 expression
The cholesterol retention in LEs/LYs not only disrupts the homeostasis of cholesterol in cells, but also impairs the fusion of APs with LYs to form autolysosomes, resulting in a defective autophagic flux. It is known that autophagy plays an important role in the drug resistance of cancer cells. Therefore, we wondered whether the autophagy is affected after the knockdown of ARL4C. Knockdown of ARL4C had minor effect on ULK1, an important factor for the formation autophagosome (Figure 6a). The early autophagy is probably not affected by the knockdown of ARL4C. However, the LC3II/LC3I ratio and p62 protein level were increased after the knockdown of ARL4C, but OSBPL5 overexpression suppressed the increase in LC3II/LC3I ratio and p62 protein. We hypothesized that the autophagy flux in later autophagy is blocked, which results in the increase of the LC3II/LC3I ratio and p62 protein level. To verify the hypothesis, we transfected cells with LC3-GFP-mRFP to evaluate the function of autophagy flux. GFP is vulnerable to the acid environment in LYs. If APs enter into LYs, GFP in LC3-GFP-mRFP is degraded with the reduction of green fluorescence. By the contrast, mRFP is more stable than GFP to the acid environment in LYs, therefore the red fluorescence of mRFP is rarely affected. If autophagosomes fail to enter into LYs, both the green and red fluorescence is rarely affected. Knockdown of ARL4C increased the green fluorescence in cells compared to the control group, but OSBPL5 overexpression suppressed the increase of green fluorescence. These results suggested that ARL4C depletion impaired the fusion of APs and LYs by suppressing OSBPL5 expression. ARL4C depletion impaired the autophagy by suppressing OSBPL5 expression. OVCAR3(R) and SKOV3(R) were subjected to treatments with ARL4C knockdown alone or in combination with OSBPL5 overexpression. (a) Western blot analysis evaluated autophagy-related proteins; ***p < 0.001 vs. Carboplatin treatment alone; ##p < 0.01 and ###p < 0.01 vs. Carboplatin treatment with ARL4C knockdown. (b) The fluorescence of LC3-GFP-mRFP was used to evaluate the function of autophagy flux in OVCAR3(R) and SKOV3(R).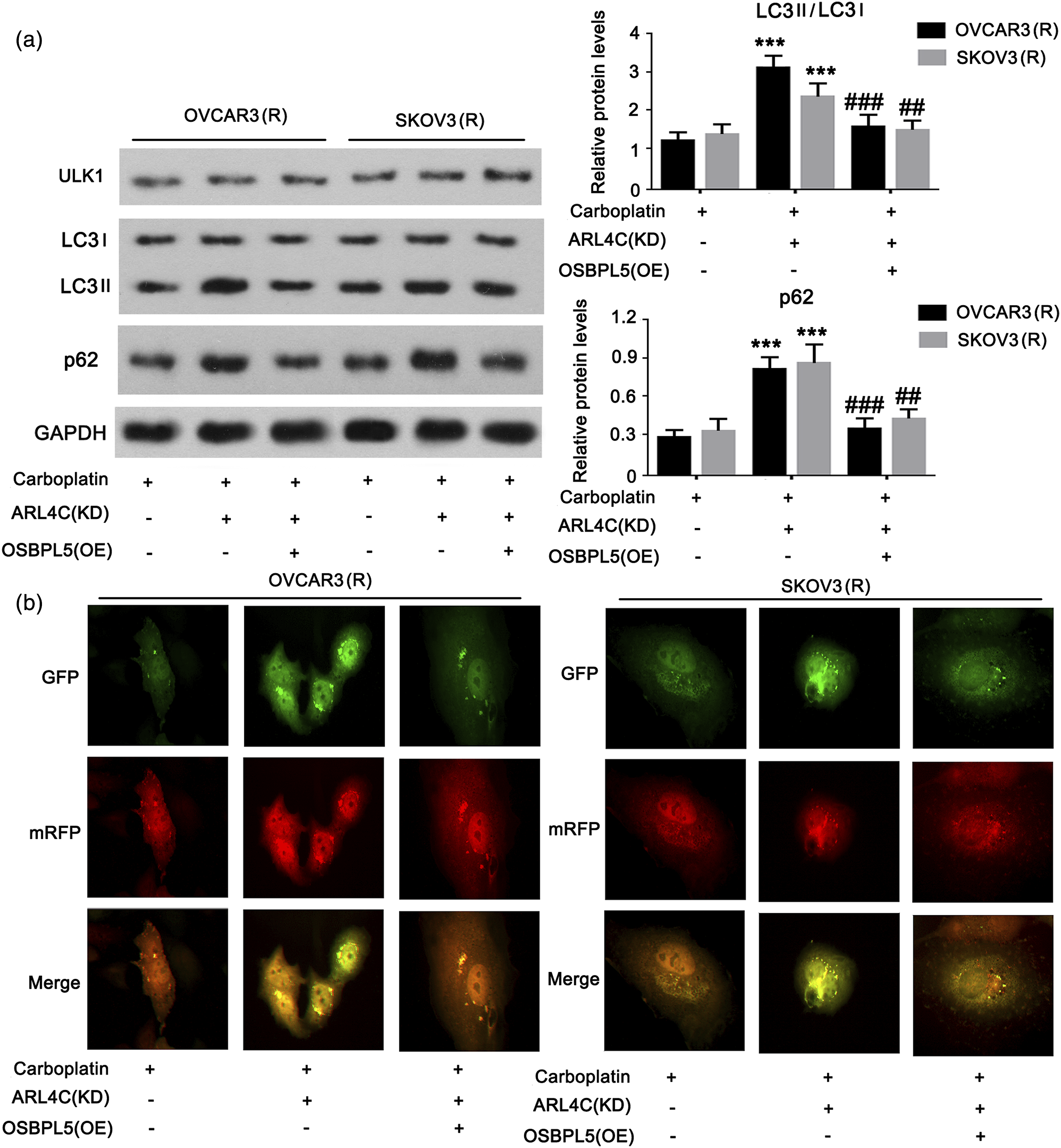
The regulatory effect of ARL4C on OSBPL5 is associated to Notch-RBP-Jκ-KDM6B pathway
High-throughput sequencing together with GO analysis suggested that Notch signal is significantly changed after ARL4C knockdown. RBP-Jκ is the primary transcript downstream of Notch signal. RBP-Jκ is activated when binding to Notch intracellular domain, NICD. RBP-Jκ transcriptional activates the target genes such as Hey1 and Hes1, as well as functions as an important regulator of histone methylation partially by KDM6B. Bioinformatics analysis showed that OSBPL5 is not only the target of RBP-Jκ (Figure 7a–c), but also the expression of OSBPL5 is influenced by histone 3 lysine 4 trimethylation (H3K4Me3) (Figure 7d). Therefore, we hypothesized that the regulatory effect of ARL4C on OSBPL5 is associated to Notch-RBP-Jκ-KDM6B pathway. As indicated by western blot, ARL4C knockdown resulted in the reduction of NICD1, NICD4, Hey1 and Hes1, suggesting the suppression of the Notch signal (Figure 8a). Co-IP analysis further confirmed the suppressed Notch signal, because the abundance of NICD4 connecting to RBP-Jκ was reduced with the ARL4C knockdown. To confirm that OSBPL5 is regulated by RBP-Jκ, we knocked down RBP-Jκ. The depletion of RBP-Jκ suppressed mRNA and protein levels of OSBPL5 (Figure 8b and c). Silencing RBP-Jκ increased KDM6B protein level, which is consistent with the result in previous report.
16
Luciferase report assay was performed to verify OSBPL5 is transcriptionally regulated by RBP-Jκ. Overexpression of RBP-Jκ increased the luciferase activity of OSBPL5-WT, but had minor effect on OSBPL5-MT (Figure 8d). We conducted ChIP assay in which anti-H3K4Me3 antibody was used to precipitate H3K4Me3 protein and PCR was performed to detect the enrichment of OSBPL5 gene promoter on H3K4Me3 protein. Both ARL4C and RBP-Jκ knockdown decreased the enrichment of OSBPL5 gene promoter on H3K4Me3 protein (p < 0.001). Bioinformatics analysis of the regulatory effect of RBP-Jκ on ARL4C. (a) Jaspar analysis shows that OSBPL5 is a target of RBP-Jκ; (b) the DNA-binding motif of RBP-Jκ; (c) the correlation between OSBPL5 and RBP-Jκ in OVC, as indicated by data from TCGA; (d) The expression of OSBPL5 is influenced by histone 3 lysine 4 trimethylation (H3K4Me3), as indicated by Genome Browser Gateway (http://www.genome.ucsc.edu/cgi-bin/hgGateway?redirect=manual&source=www.genome.ucsc.edu). The regulatory effect of ARL4C on OSBPL5 is associated to Notch-RBP-Jκ-KDM6B pathway. (a) Western blot evaluated NICD1, NICD4, Hey1 and Hes1 after ARL4C knockdown. In addition, Co-IP analysis further analyzed the abundance of NICD4 on RBP-Jκ after ARL4C knockdown. RBP-Jκ was knocked down in OVCAR3(R) and SKOV3(R), followed by (b) PCR and (c) western blot assays. (d) Luciferase report assay was performed to verify OSBPL5 is transcriptionally regulated by RBP-Jκ. (e) We conducted ChIP assay in which anti-H3K4Me3 antibody was used to precipitate H3K4Me3 protein and PCR was performed to detect the enrichment of OSBPL5 gene promoter on H3K4Me3 protein. ***p < 0.001 vs. Control.

Discussion
This study found that ARL4C is up-regulated in OVC that resistant to Carboplatin, while ARL4C knockdown increased the sensitivity of OVC to Carboplatin. Wakinoue et al. found that more than half patients with endometriosis-associated ovarian cancer have high level of ARL4C. 10 Moreover, higher level of ARL4C is associated to worse 5-year overall survival. 10 ARL4C, hence, is a promising therapeutic target in EAOC or other types of OVC. As ARL4C is closely associated to the export of cholesterol with ABCA1, we tested the regulatory effect of ARL4C on cholesterol in OVC cells. ARL4C knockdown led to the increase of free cholesterol in Carboplatin-resistant OVC, probably due to the loss of the function of cholesterol export. ABCA1 is also important for cholesterol export. It has been reported that inhibition of ABCA1 suppresses the growth and migration of epithelial ovarian cancer. 17 However, in this study, ABCA1 overexpression failed to reverse the increase of free cholesterol caused by ARL4C knockdown. This suggests that ABCA1 is unable to replace the role of ARL4C in the export of cholesterol. The dysfunction of cholesterol export probably results in the accumulation of cholesterol in OVC. However, the increase of cholesterol in OVC is generally associated with enhanced drug resistance.18,19 It seems to be contradictory to the increased sensitivity of OVC to Carboplatin with ARL4C knockdown.
This study found that ARL4C not only participates in the cholesterol export, but also influences cholesterol import by regulating OSBPL5. Cholesterol import is linked to the uptake of cholesterol to LYs. As LYs are unable to store cholesterol, it is further transported to ER for Cholesterol esterification, which is the primary approach for cholesterol storage in cells. We found that ARL4C knockdown impaired the transport of cholesterol from LYs to ER in OVC cells due to the reduction of radio-labeled cholesterol esters in the cholesterol esterification assay. Interestingly, cholesterol esters had no significant change after ARL4C knockdown. It is probably because the blockage of cholesterol export by ARL4C knockdown increases cholesterol esters in ER, which offsets the reduction of newly formed cholesterol esters in ER. OSBPL5 overexpression recovered the transport of cholesterol from LYs to ER in OVC cells after ARL4C knockdown. This suggests that the effect of ARL4C on the transport of cholesterol from LY to ER is dependent on OSBPL5. Study has confirmed that OSBPL5 depletion leads to the retention of cholesterol in LYs. 15 The cholesterol retention in LYs probably leads to the dysfunction of autophagy.
Numerous evidences indicate autophagy contributes to the drug resistance of cancer cells. 20 This study for the first reported that ARL4C is implicated in the regulation of autophagy. Silencing ARL4C suppressed the fusion of APs with LYs to form autolysosomes, suggesting the dysfunction of autophagy. ARL4C knockdown had minor effect on ULK1, but increased the LC3II/LC3I ratio and p62 protein level. These results suggested that ARL4C depletion did not affect the early autophagy (the formation of APs) but disrupted the later autophagy (the fusion of APs with LYs). Overexpression of OSBPL5 recovered the autophagy in OVC after ARL4C knockdown. Therefore, the regulatory effect of ARL4C knockdown on autophagy is associated to the reduction of OSBPL5. Shang et al. reported that OSBPL5 deficiency is associated with inhibited autophagy in transient cerebral ischemia. 21 This suggests that OSBPL5 is important for the normal function of autophagy. The resistance of OVCAR3(R) and SKOV3(R) to Carboplatin was attenuated with ARL4C depletion. However, the resistance was restored when autophagy was recovered by OSBPL5 overexpression.
This study revealed that the regulatory effect of ARL4C on OVC resistance to Carboplatin is closely associated to OSBPL5. Therefore, we were curious about the mechanism by which ARL4C regulates OSBPL5. High-throughput sequencing together with GO analysis suggests that Notch is the downstream signal of ARL4C. When activated, Notch protein released NICD to stimulate the transcriptional function of RBP-Jκ. We confirmed that RBP-Jκ transcriptionally regulates OSBPL5 by binding to the promoter region. RBP-Jκ also functions as an important regulator of histone methylation partially by KDM6B. 16 RBP-Jκ suppresses KDM6B, leading to increased H3K4Me3. H3K4Me3 facilitates gene expression,22,23 which probably enhances the transcription of OSBPL5 by RBP-Jκ.
In summary, ARL4C is up-regulated in OVC that resistant to Carboplatin, while ARL4C knockdown increased the sensitivity of OVC to Carboplatin. As shown in the mechanism diagram (Figure 9), ARL4C knockdown led to the reduction of OSBPL5 by suppressing Notch-RBP-Jκ-H3K4Me3. OSBPL5 deficiency inhibited the transport of cholesterol from LYs to ER, which led to the accumulation of cholesterol in LYs and the dysfunction of autophagy. Therefore, ARL4C knockdown attenuated the resistance of OVC to Carboplatin by disrupting cholesterol transport and autophagy. This study revealed a promising target to attenuate the resistance of OVC to Carboplatin and elucidated the potential mechanism. The mechanism diagram of the effect of ARL4C on the resistance of OVC to Carboplatin. By suppressing Notch signal, ARL4C knockdown inhibited the transcriptional function of RBP-Jκ and RBP-Jκ-induced H3K4Me3, which collectively reduced OSBPL5 expression. OSBPL5 deficiency inhibited the transport of cholesterol from LYs to ER, which led to the accumulation of cholesterol in LYs and the dysfunction of autophagy. Therefore, ARL4C knockdown attenuated the resistance of OVC to Carboplatin by disrupting cholesterol transport and autophagy.
Footnotes
Author contributions
JY and SP conceived and designed the study. JY, KZ, and SP performed experiments and interpreted the data. JY collected the data. KZ and SP analyzed the data and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Hunan Cancer Hospital Climb Plan.
Ethics approval
This study was approved by the Ethics Committee of Hunan Cancer Hospital and the Affiliated Cancer Hospital of Xiangya School of Medicine, and all patients involved provided written informed consent.
Consent for publication
Written informed consent for publication was obtained from all participants.
Data availability
The datasets generated/analyzed in the present study are available upon reasonable request from the corresponding author.