
Abstract
Treatment with aluminum chloride (AlCl3) suppresses the growth of osteoblastic cells; however, the molecular mechanisms underlying the impact of AlCl3 on cell growth have not been fully characterized. In this study, we observed that exposure of hFOB1.19 cells to AlCl3 arrested cells at G0/G1 phase by inducing p21 expression. Further studies indicated that AlCl3 upregulated the phosphorylation level of signal transducer and activator of transcription 1 (STAT1) at serine 727 site (Ser727). By chromatin immunoprecipitation and electrophoretic mobility shift assay, we found that AlCl3 promoted STAT1/DNA binding activity to p21 promoter, thus resulting in the upregulation of p21. Moreover, siRNA-mediated knockdown of STAT1 attenuated p21 level induced by AlCl3. Notably, using hFOB1.19 cells stably expressing dominant-negative STAT1 (Ser727Ala), we demonstrated that phosphorylation of STAT1 at Ser727 site is required for p21-mediated cycle arrest induced by AlCl3. Mechanism investigation indicated that AlCl3 stimulated the phosphorylation of JNK, and administration of JNK inhibitor SP600125 prevented AlCl3-induced G0/G1 arrest through suppressing the phosphorylation of STAT1. Notably, pretreatment with N-acetyl-cysteine, a reactive oxygen species scavenger, conferred a significantly inhibitory effect on AlCl3-mediated activation of JNK/STAT1 signaling pathway. Taken together, our findings provide the molecular mechanism for G0/G1 arrest induced by AlCl3 in osteoblastic cells.
Introduction
Aluminum (Al) can enter into and accumulates in humans through various sources such as diet, air, drinking water, cosmetics, and medication. Accumulating evidence showed that excessive Al exposure causes a variety of adverse effects on hard and soft tissues.1,2 Indeed, previous studies have reported that excessive Al accumulation inhibits osteoblasts proliferation and differentiation.2,3 Al-induced reduction in osteoblastic bone formation results in deteriorated bone architecture and decreased bone mass.4,5 However, the underlying molecular mechanism responsible for the reduced cell proliferation of osteoblasts during Al exposure remains largely unknown.
As an essential and fundamental biological process, cell division exists in all multicellular organisms. 6 The cell cycle consists of four phases and is a sequence of events that leads to cell division. 7 Excessive Al exposure could decrease cell proliferation, which was reflected in the cell cycle processes and the expression of regulators involved in cell cycle progression. 8 Recent research indicated that Al-mediated suppression of cell growth correlates with cell cycle arrest. 9 However, the potential effects of Al on cell cycle progression of osteoblasts and the underlying mechanisms by which Al induced cell cycle arrest have not been adequately investigated.
The signal transducer and activator of transcription (STAT) factors is a family member of cytoplasmic proteins with roles as transcription factors and signal messengers that induced by a wide variety of cytokines and growth factors. 10 The STAT protein family is composed of seven members, though some of which exist in different isomeric forms, all of their structural domains are highly conserved. These contain a coiled-coil domain, an amino-terminal domain, an SH2 domain, a DNA-binding domain, and a transactivation domain. The transactivation domain contains one or two amino acid residues which are pivotal for the STAT activity. 11 STAT1 has been implicated in regulating cell proliferation, both negatively and positively, through down- or up-regulation cell cycle regulators.12,13 When STAT1 was activated by phosphorylation, and subsequently translocate to the cell nucleus, where they transactivate the responsive genes. 14 Hence, this prompted us to examine whether STAT1 plays a role in cell cycle arrest in the presence of Al. In the current study, we aim to explore the underlying mechanism of aluminum chloride (AlCl3) inhibitory effect on cell growth and the involvement of STAT1 signaling in osteoblasts.
Materials and methods
Reagents and antibodies
AlCl3 (563,919) and N-acetyl-cysteine (NAC, A9165) were obtained from Sigma-Aldrich (St Louis, MO, USA). The following antibodies, anti-p21 (2947, 1:1000 dilution); anti-p27 (3686, 1:1000 dilution), anti-p53 (2527, 1:1000 dilution), anti-p38 (8690, 1:1000 dilution), anti-p-p38 (4511, 1:1000 dilution), anti-AKT (4685, 1:1000 dilution), anti-p-AKT (4060, 1:1000 dilution), anti-ERK (4695, 1:1000 dilution), anti-p-ERK (4370, 1:1000 dilution), anti-JNK1/2 (9252, 1:1000 dilution), anti-p-JNK1/2 (9255, 1:1000 dilution), anti-β-actin (4970, 1:3000 dilution), and Horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (14,708, 1:1000 dilution) were obtained from Cell Signaling Technology (Beverly, MA, USA); anti-STAT1 (ab234400, 1:1000 dilution), anti-phospho-STAT1 (Ser727, ab109461, 1:1000 dilution), anti-phospho-STAT1 (Tyr701, ab109457, 1:1000 dilution), and anti-Histone H3 (ab1791, 1:2000 dilution) were from Abcam (Cambridge, UK). Polyvinylidene difluoride (PVDF, ISEQ00005) membrane was obtained from Millipore (Bedford, MA, USA). Enhancer chemiluminescent (ECL) reagent (A38555) was purchased from Pierce Biotechnology (Rockford, IL, USA). The specific JNK inhibitor SP600125 (S1460) was obtained from Selleck Chemicals (Houston, TX, USA) and was dissolved in dimethyl sulfoxide (DMSO). Polybrene (GM-040,901) was obtained from Genomeditech Co. Ltd (Shanghai, China) and was used at the indicated concentration. The standard stock and trial solutions were prepared in strict according to the manufacturer’s instructions.
Cell culture
The human osteoblast cell line hFOB1.19, purchased form ATCC (Manassas, VA, USA), were maintained in DMEM/F-12 medium (GIBCO, Grand Island, NY, USA) containing 10% fetal bovine serum (Hyclone, Logan, UT, USA), geneticin (400 μg/mL, Sigma, St Louis, MO, USA), and 1% penicillin/streptomycin. The hFOB1.19 cells were cultured in a 5% CO2-humidified atmosphere at 37°C.
Experimental protocols
Step 1: Determination the effects of AlCl3 on cell growth. The hFOB1.19 cells were treated with different concentrations of AlCl3 (0, 0.5, 1, 2, and 4 mM) for 24 and 48 h, respectively.
Step 2: Determination the role of p21 in AlCl3-induced cell cycle arrest. (1) The cells were treated with 0, 0.5, 1, and 2 mM of AlCl3 for 48 h. (2) The hFOB1.19 cells were randomly divided into four groups: Scramble siRNA, Scramble siRNA + AlCl3 (2 mM), p21 siRNA, and p21 siRNA + AlCl3 (2 mM).
Step 3: Determination the effects of JNK/STAT1 signaling in AlCl3-induced p21 upregulation. (1) Cells were treated with AlCl3 (0, 0.5, 1, and 2 mM) for 48 h. (2) The hFOB1.19 cells were randomly divided into four groups: Scramble siRNA, Scramble siRNA+AlCl3 (2 mM), STAT1 siRNA, and STAT1 siRNA+AlCl3 (2 mM). (3) The hFOB1.19 cells were randomly divided into four groups: Control, AlCl3 (2 mM), STAT1-S727 A, and STAT1-S727 A + AlCl3 (2 mM). (4) The cells were randomly divided into four groups: Control, AlCl3 (2 mM), SP600125 (20 μM), and SP600125 (20 μM) + AlCl3 (2 mM).
Step 4: Determination the role of ROS in AlCl3-induced JNK/STAT1/p21 siganling activation. The cells were randomly divided into four groups: Control, AlCl3 (2 mM), NAC (5 mM), NAC (5 mM) + AlCl3 (2 mM).
Cell viability assay and viable cell counts
The Cell Counting Kit-8 (CCK-8) assay kit (Dojindo Laboratories, Kumamoto, Japan) was used to evaluate the cell viability. In brief, a total of 5 × 103 cells/well were seeded into 96-well plates. After treatment, 10 μL of CCK-8 reagent (the ratio of medium to CCK-8 reagent is 10∶1) was added into each well and incubated for 2 h away from light. The absorption was measured at 450 nm by a microplate spectrophotometer (PerkinElmer, Waltham, MA, USA).
For viable cell counts, hFOB1.19 cells were stained with 0.4% Trypan blue (Beyotime, Shanghai, China) to distinguish the fractions of dead and live cells. The number of Trypan blue-positive and excluded Trypan blue cells was counted in a hemocytometer.
Cell cycle analysis
For cell cycle assay, control and treated cells were collected into flow cytometry tubes and centrifuged at 2000 r/min for 5 min to obtain cell pellets. The cells were washed with PBS and fixed with 70% ethanol at 4°C overnight, and stained with propidium iodide (PI). The distribution of cell cycle was analyzed by flow cytometry (Beckman-Coulter, Miami, FL, USA).
SiRNA transfection
For siRNA knockdown, hFOB1.19 cells were transfected with siRNA specific for p21, p53, STAT1, or scramble siRNA (Thermo Fisher Scientific, Carlsbad, CA, USA) using Lipofectamine RNAi/MAX Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. After 24 h of incubation, the cells were then stimulated with AlCl3 (0 or 2 mM) for 48 h and harvested for further experiments.
Virus production and lentiviral transduction
To confirm the STAT1 function, lentivirus expression vectors were introduced. The pLV-S727A-STAT1 vector was obtained from Addgene (Cambridge, MA, USA). In brief, the S727 A lentivirus vector was co-transfected into 293T packaging cells with the packaging plasmids psPAX2 and pMD2.G (Addgene) in the presence of Lipofectamine 3000 transfection reagent (Invitrogen) to generate lentivirus. After 24 h and 48 h of transfection, the supernatant was collected, centrifuged, and then filtered with a 0.4-μm filter. The hFOB1.19 cells were transduced with lentiviral vector particles in the presence of Polybrene (6 μg/mL) for 24 h. To select stably transfected cells, cells were treated with 4 μg/mL puromycin (Thermo Fisher) for 12–14 days.
Western blotting
Nuclear extraction kit (Beyotime Shanghai, China) was used to isolate nuclear fractions. The whole cell lysate was lysed with radioimmunoprecipitation (RIPA) buffer. Equal amounts of proteins were resolved on SDS-PAGE (8–12%) and transferred onto PVDF membranes. Membranes were incubated with either 5% skim milk or bovine serum albumin (BSA/TBST) and probed with specific primary antibodies overnight at 4°C on a shaker, and then incubated with secondary antibodies for 1 h at room temperature. The signal bands were visualized via chemiluminescence detection reagent.
Reverse transcription quantitative polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen), and cDNA was synthesized from total RNA (2 μg) by using the Moloney-murine leukemia virus reverse transcriptase (Promega, Madison, WI, USA) according to the manuscript’s protocols. RT-qPCR was performed on ABI StepOnePlus PCR system (Applied Biosystems, Foster City, CA, USA) using the SYBR Premix ExTaq II (Takara, Otsu, Japan). The primer pairs used for RT-qPCR were as follows: p21 forward, 5′-TGTCCGTCAGAACCCATGC-3′, p21 reverse; 5′-AAAGTCGAAGTTCCATCGCTC-3′; β-actin forward, 5′-CATGTACGTTGCTATCCAGGC -3′, β-actin reverse, 5′-CTCCTTAATGTCACGCACGAT-3′.
Chromatin immunoprecipitation assay
A ChIP assay was performed with the Pierce Agarose ChIP Kit (Thermo Fisher Scientific). In brief, hFOB1.19 cells were cross-linked in 1% formaldehyde and quenched with 1.25 mM glycine. Cell lysates were then collected with cold lysis buffer for ChIP and sonicated under optimized conditions. The chromatin samples were immunoprecipitated with STAT1-ChIP grade antibody. The immuoprecipitates were then incubated with protein A/G agarose beads for 2 h. The protein-DNA complex was eluted with elution buffer containing proteinase K and the cross-links were reversed at 65°C for 12 h. DNA was extracted with phenol-chloroform and was analyzed by real-time PCR. The human p21 promoter primers used for ChIP assay was purchased from Cell signaling technology.
Electrophoretic mobility shift assay
The STAT1/DNA-binding activity was performed with an Electrophoretic Mobility Shift Assay Kit (Molecular Probes, Invitrogen, CA, USA) according to the manufacturer’s recommendations. The probe sequence for STAT1 is as follows: 5′-CATGTTATGCATATTCCTGTAAGTG-3’ (sense strand). The protein-DNA complex was applied to 6% non-denaturing PAGE.
Measurement of intracellular reactive oxygen species
Intracellular ROS level was measured with a ROS assay kit (Beyotime) that sets DCFH-DA as the probe. The intensity of the DCF signal were detected by flow cytometry to quantify the intracellular production of ROS. The hFOB1.19 Cells were incubated with 10 μM DCFH-DA-containing serum-free medium for 20 min, and analyzed by a flow cytometer.
Statistical analysis
The analyses were performed using the SPSS19.0 software (Chicago, IL, USA). All values are presented as means ± standard error of the mean (SEM). Data of three independent experiments unless otherwise specified. Statistical differences were determined by Student’s t-test between two groups. Statistical differences were calculated using ANOVA followed by Bonferroni post hoc test in multiple groups. A p value less than 0.05 was considered significant.
Results
AlCl3 induces p21-dependent G0/G1-phase arrest in hFOB1.19 cells
Firstly, to verify whether AlCl3 inhibits the cell growth of hFOB1.19 cells, we investigated the effect of AlCl3 on cell viability in hFOB1.19 cells and found that AlCl3 could inhibit cell viability of hFOB1.19 cells in a dose- and time-dependent manner (Figure 1(a)). By using Trypan blue staining assay, the effect of AlCl3 on cell death in hFOB1.19 cells was studied. The cell death ratio elicited by 0.5–2 mM AlCl3 exposure did not correspond to the decreased cell viability under the identical dosage of AlCl3 treatment (Figure 1(b)). Thus, AlCl3 mainly inhibited cell growth without causing significant cell death at doses less than 2 mM. AlCl3 treatment inhibits the growth of hFOB1.19 cells. Cells were treated with 0, 0.5, 1, 2, and 4 mM of AlCl3 for 24 and 48 h. (a) Growth inhibition of hFOB1.19 cells was assessed by CCK-8 assay. (b) The death rate of hFOB1.19 cells was determined by survival cell count assay. The data was presented as the mean ± SEM (n = 3). *p < 0.05 and **p < 0.01.
Next, to investigate the mechanism underlying the inhibition of cell growth induced by AlCl3, we analyzed the effects of AlCl3 on the cell cycle phase distribution by flow cytometry after PI staining. As shown in Figure 2(a) and (b), AlCl3 induced the cell cycle arrest at G0/G1 phase. The percentage of cells in the G0/G1 phase increased from 45.72% (control) to 71.75% (2 mM) in hFOB1.19 cells. Moreover, AlCl3 treatment increased p21 expression, but had no regulatory effect on p27 expression (Figure 2(c)). To further characterize the functional role p21 in AlCl3-induced cell cycle arrest, we silenced the expression of p21 and confirmed the reduction of p21 expression by western blot (Figure 2(d)). As shown in Figure 2(e) and (f), p21-siRNA reversed the cell cycle arrest induced by AlCl3. The data confirmed that p21 has an important role in AlCl3-induced G0/G1 phase arrest. AlCl3 treatment induced G0/G1 phase arrest that was dependent on p21 level in hFOB1.19 cells. (a and b) Cells were treated with 0, 0.5, 1, and 2 mM of AlCl3 for 48 h, and then the cell cycle distributions were analyzed by flow cytometry. (c) Cells were treated with various concentrations of AlCl3 for 48 h. The levels of p21 and p27 were determined by western blot. Cells were transfected with scramble or p21 siRNA for 24 h, followed by treated with 2 mM AlCl3 for 48 h. The p21 level (d) and percentage of cell cycle distribution (e and f) were determined. The data was presented as the mean ± SEM (n = 3). **p < 0.01.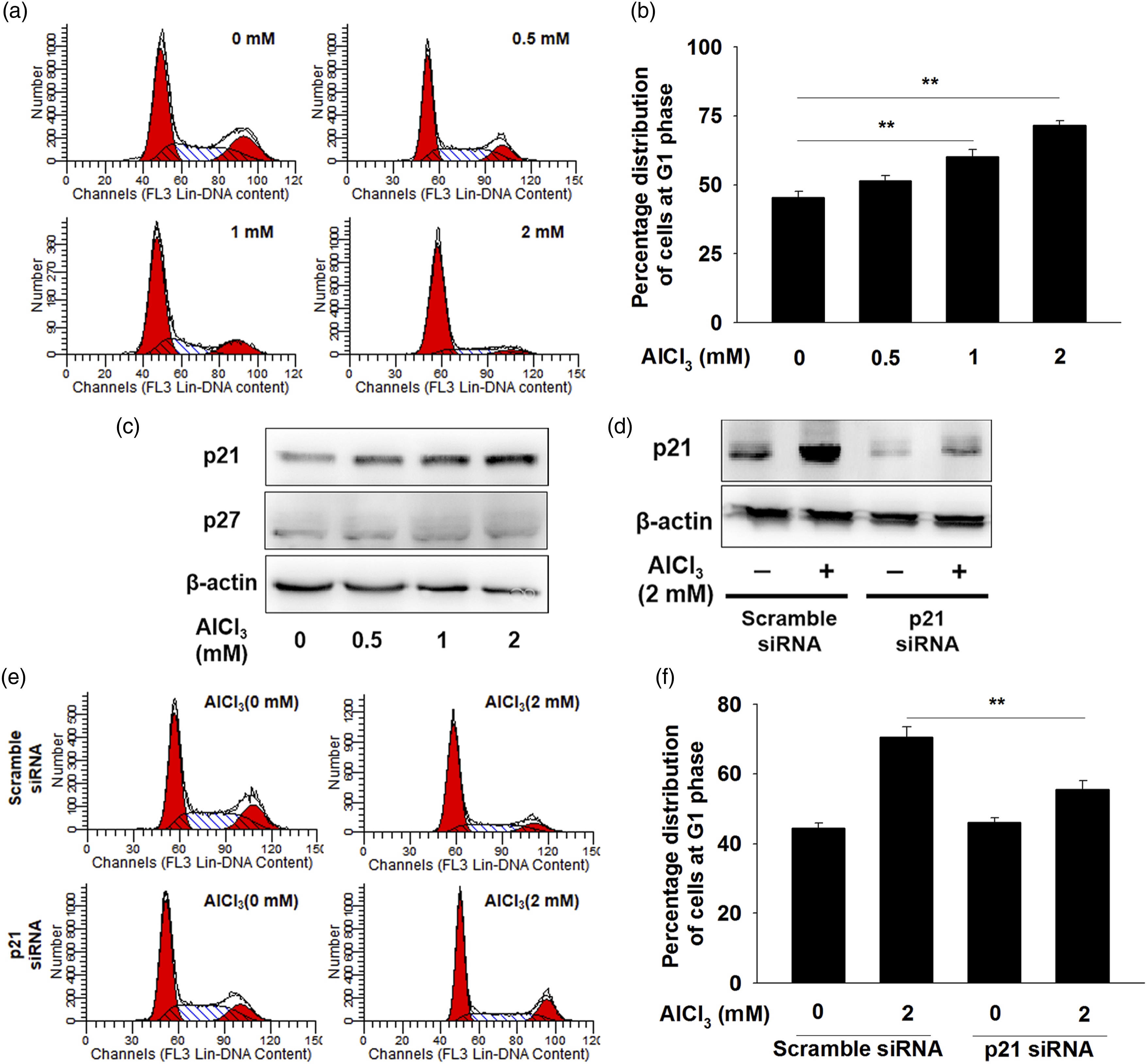
Role of STAT1 in AlCl3-mediated p21 expression in hFOB1.19 cells
Previous researches showed that p21 could be regulated by p53.
15
We therefore sought to establish whether p53 regulated the p21 expression. As shown in Figure 3(a), the results showed that the level of p53 increased significantly after treatment with AlCl3. However, silencing p53 by specific siRNA had no effects on p21 expression in hFOB1.19 cells (Figure 3(a)). STAT1 is involved in AlCl3-induced G0/G1 arrest in hFOB1.19 cells. (a) Cells were transfected with scramble or p53 siRNA for 24 h and then treated with 2 mM AlCl3 for 48 h. Cell lysates were harvested and analyzed of the p21 expression by western blot. (b) Cells were treated with or without AlCl3, and then the whole-cell extracts were prepared and subjected to western blot using the indicated antibodies. (c) The nuclear extracts were prepared and analyzed for the levels of phosphorylated STAT1 and total STAT1. The hFOB1.19 cells were transfected with scrambled siRNA or STAT1-siRNA, followed by AlCl3 treatment, and then the changes of STAT1 expression (d) and cell cycle distribution (e and f) were determined. The data was expressed as the mean ± SEM (n = 3). **p < 0.01.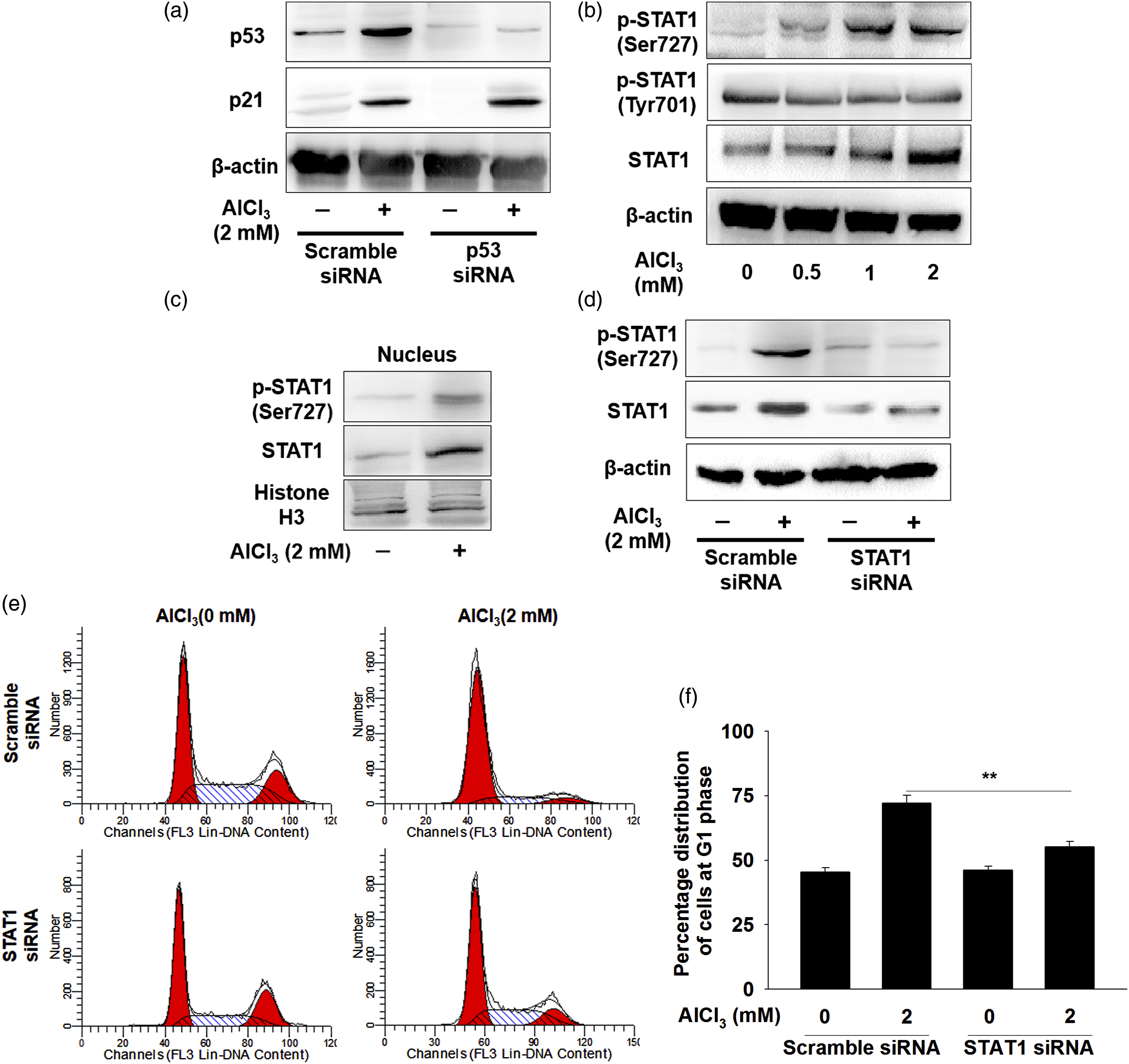
As the stress-activated transcription factor, STAT1 signaling plays an important role in cell cycle arrest. The influence of AlCl3 on STAT1 signaling was observed by using immunoblotting. As shown in Figure 3(b), the enhanced phosphorylation of STAT1 at Ser727 and total STAT1, not Tyr701, were observed in cells treated stimulated with AlCl3. Since STAT1 was activated by phosphorylation, and then translocate to the cell nucleus to function as transcription regulators. As shown in Figure 3(c), the unclear extracts also exhibited increased STAT1 Ser727 level under AlCl3 exposure. Given that STAT1 signaling was activated, we further evaluated the possibility that AlCl3 induces cell cycle arrest depending on the STAT1. As shown in Figure 3(d) to (f), knockdown of STAT1 inhibited the phosphorylation of STAT1 and also eliminated the capacity of AlCl3 to induce G0/G1 arrest. These results indicated that STAT1 was essential for AlCl3-induced G0/G1 arrest.
To explore the role of STAT1 in p21 expression regulation, we performed the ChIP assay and found that the relative binding of STAT1 to the p21 promoter was markedly increased in response to AlCl3 treatment (Figure 4(a)). In addition, the results of the EMSA also demonstrated that AlCl3 exposure increased the STAT1/DNA-binding activity (Figure 4(b)). As shown in Figure 4(c) and (d), the increased on the mRNA and protein levels of p21 induced by AlCl3 were attenuated in cells transfected with STAT1 siRNA, while the scramble siRNA had no effect on p21 expression in hFOB1.19 cells treated with AlCl3. AlCl3 stimulates p21 expression via STAT1 signaling in hFOB1.19 cells. (a) Cells were treated with 2 mM AlCl3 for 48 h, and the in vivo binding of STAT1 to p21 promoter was examined by ChIP assay. Fold-enhancement was determined by normalizing threshold cycle values of STAT1 ChIP against IgG ChIP. (b) An EMSA assay was performed to analyze STAT1/DNA-binding activity. (c) and (d) Cells were transfected with scramble siRNA or STAT1 siRNA before AlCl3 exposure, then the mRNA and protein levels of p21 were determined. (e) The protein level of p21 in S727A-overexpressed cells were determined after AlCl3 stimulation by western blot analysis. (f and g) The distribution of cell cycle was analysed by PI staining in STAT1 phospho-mutant (S727 A) cells treated with AlCl3. Each point represents the mean ± SEM (n = 3). *p < 0.05.
To further shed further light on the role of STAT1 activation in AlCl3-induced cell cycle arrest, STAT1 phosphorylation mutant (STAT1 Ser727Ala) was used. As shown in Figure 4(e), the phosphorylation of STAT1 at Ser727 plays a key role as a prerequisite for AlCl3-induced up-regulation of p21. This phospho-mutant led to an escape from AlCl3-induced G0/G1 arrest (Figure 4(f) and (g)). Collectively, all these data clearly indicating that STAT1 phosphorylation (Ser727) was essential for AlCl3-induced p21 expression.
STAT1 activation induced by AlCl3 is dependent on JNK pathway
Previous studies revealed that serine phosphorylation of STAT1 could be regulated by MAPK or PI3K-AKT pathway.16–18 As shown in Figure 5(a), phosphorylation of AKT was abolished by AlCl3 treatment, while AlCl3 exhibited little effects on the phosphorylation of p38 and ERK. In contrast, the phosphorylation level of JNK1/2 was markedly increased after AlCl3 treatment (Figure 5(a)). Western blotting analysis demonstrated that SP600125 inhibited AlCl3-induced serine phosphorylation of STAT1 (Figure 5(b)). Additionally, gel shift analysis revealed that treatment with SP600125 decreased the STAT1/DNA-binding activity in AlCl3-treated hFOB1.19 cells (Figure 5(c)). Collectively, our data indicated that AlCl3-induced activation of STAT1 was dependent on JNK signaling. STAT1 activation induced by AlCl3 is dependent on JNK pathway. (a) hFOB1.19 cells were treated with AlCl3 (2 mM) for 48 h. The expressions of phosphorylated AKT, ERK, p38, and JNK as well as their total proteins were analyzed by western blotting. (b) The hFOB1.19 cells were pretreated with 20 μM SP600125, followed by AlCl3 treatment. The p-JNK level, p-STAT1 (Ser727), and p21 levels were determined. (c) The nuclear extracts were prepared and the STAT1/DNA binding were studied.
ROS generation is upstream of JNK/STAT1/p21 signaling
An increasing number of studies have shown that AlCl3 exposure results in the excessive production of ROS. We detected the intracellular ROS level and found that treatment with AlCl3 significantly increased the ROS production in hFOB1.19 cells. Preincubation of cells with scavenger NAC markedly blocked AlCl3-induced ROS (Figure 6(a) and (b)). Moreover, NAC could markedly reverse AlCl3-induced cell cycle arrest (Figure 6(c) and (d)). We further explored the relationship between ROS generation and JNK activation in AlCl3-treated hFOB1.19 cells. Western blot analysis revealed that pretreated with NAC could prevent the upregulation of JNK and STAT1 phosphorylation in AlCl3-treated hFOB1.19 cells (Figure 6(e)). Moreover, the increased p21 expression were reserved by ROS scavenger treatments (Figure 6(e)). Taken together, these data indicated that AlCl3-induced ROS production contributed to the activation of the JNK/STAT1/p21 signaling. ROS generation is upstream of the JNK/STAT1 signaling pathway. hFOB1.19 cells were pre-treated with NAC (5 mM) for 2 h, and then treated with AlCl3 (2 mM) for additional 48 h. (a) and (b) The ROS level was determined. Histograms indicate the fold change in DCFH-DA intensity. (c) and (d) The cell cycle was analyzed by flow cytometry. (e) Cells were treated with 2 mM AlCl3 after pretreatment with 5 mM NAC for 2 h. Whole-cell extracts were prepared and subjected to western blot using the indicated antibodies. Results are expressed mean ± SEM (n = 3). **p < 0.01.
Discussion
The cell cycle is a set of monitored and organized events responsible of cell division into two daughter cells, and normal cell cycle progression is necessary for the maintenance of cell growth homeostasis in proliferating cells. 19 Although some previous studies have suggested AlCl3 could inhibit the cell growth of osteoblastic cells, 20 the molecular mechanism underlying their action remains to be fully elucidated. In this study, we reported that AlCl3 arrests the osteoblastic cells at the G0/G1 phase. Cell cycle progression is regulated by a wide variety of external factors, in which ROS may play a crucial role. The role of ROS as the intracellular messengers regulating the duration and intensity of cell signaling pathways are widely recognized. 21 Recent evidence revealed that excessive production of cellular ROS, induced by exogenous agents, will cause the activation of signaling pathways, which disturbed the progression of cell cycle.22,23 The use of the DCFH-DA probe showed that AlCl3 induced a significant increase in ROS production, while antioxidant treatment of hFOB1.19 cells reduced ROS levels and ended in cell cycle arrest in G0/G1-phase. The results of these experiments suggested that induction of ROS generation is critical in AlCl3-induced cell cycle arrest.
P21 and p27 are the key elements of the G0/G1 phase regulatory apparatus. Removal of the cyclin-dependent kinases (CDKs) inhibitory activity related to p21 and p27 in G0/G1 phase is critical to activate of cyclin-CDK complexes and induce cell-cycle progression towards S phase.24,25 From our series of studies, we found an increased level of p21 after AlCl3 treatment, but p27 level was not affected by AlCl3 treatment. Moreover, genetic knockdown of p21 prevented AlCl3-mediated G0/G1 blockade, suggesting that AlCl3 blocked the cell cycle in G0/G1 phase through upregulation of p21. Previous studies demonstrated that p53 protein acts as the main transcriptional regulator of p21. 26 Our data partially correlate with previous studies showing that AlCl3 induced p53 expression. However, it is unlikely that AlCl3 transactivated the expression of p21 in p53-dependent manner because the increased p21 expression was also detected in p53 siRNA-treated hFOB1.19 cells.
STAT proteins have been shown to promote fundamental cellular processes. The transcription factor STAT1, a member of the STAT family, is an important cytoplasmic molecule involved in cellular responses and signaling cascades initiated by cytokines and growth factors. 27 Previous research indicated that the activation of STAT1 plays a major role in the transcriptional activation p21 in a p53-independent manner. 28 However, whether AlCl3 can activate the STAT1 signaling pathway in the osteoblastic cells remains largely unclear. Our approach with siRNA clearly demonstrated that the increased p21 expression by AlCl3 is dependent on STAT1. Taken together, the results implied that the change of p21 modulated by the STAT1 gene might influence the cell cycle distribution caused by AlCl3. The activity of STAT1 is mediated by phosphorylation on Ser727 and Tyr701. Phosphorylation of STAT1 at Ser727 is required for the activation of STAT1.29,30 Given that phosphorylation is pivotal for STAT1 activation, we noted that AlCl3 treatment increased STAT1 Ser727 phosphorylation compared to the control group, but had no significant effect on STAT1 phosphorylation at Tyr701 site. Indeed, early findings have provided evidence for Ser727 phosphorylation of STAT1 is necessary for cell cycle arrest.
The impetus for this study was to investigate pathway that may be involved in promoting osteoblastic cells growth arrest under AlCl3 stimulation. Several signaling pathways have been implicated in regulating the serine phosphorylation of STAT1, including ERK, 31 JNK, 32 p38, 18 and PI3K/AKT. 16 Here we found that the phosphorylation of AKT was abrogated by AlCl3 and the phosphorylation of p38 and ERK were hardly changed after AlCl3 treatment. In contrast, JNK was significantly activated in AlCl3-treated hFOB1.19 cells, and the induction of STAT1 phosphorylation (Ser727 sites) by AlCl3 was JNK-dependent. Most importantly, we extended those observations to show that AlCl3-induced expression of JNK and its targets was fully blocked by the antioxidant NAC, indicating ROS-dependent JNK activation was critical in AlCl3-induced G0/G1 arrest.
In conclusion, we present evidence of a critical role for STAT1 in regulating AlCl3-mediated cell cycle arrest in hFOB1.19 cells initiated by ROS. For the induction of G0/G1 arrest, AlCl3 enhanced the JNK/STAT1/p21 signaling axis with STAT1 phosphorylation at Ser727 site. All these findings could provide a new clue for future research on the molecular mechanisms of AlCl3 in the field of cell growth.
Footnotes
Appendix
Authors’ contributions
YLZ and FJL conducted the experiments and data analysis. ZYL conceived and designed the study. SGL and JJJ acquired the data. JYF and WBQ performed the data analysis and interpretation. All authors have read and approved this manuscript
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Key Technologies Research and Development Program of Henan Province (132102110053). Scientific and Technological Project of Henan Province (LHGJ20200825). Science and technology program of Jiaozuo City (2020271).