
Abstract
Methamphetamine (METH) is an illicit amphetamine-like psychostimulant that is commonly abused. However, the modulation of METH-induced cardiac microvascular permeability is still not completely known. Previously, we discovered that the vascular endothelial growth factor (VEGF) regulated the cardiotoxicity produced by METH. In this work, we looked into the effect of METH exposure on cardiac microvascular permeability via the VEGF-PI3K-Akt-eNOS signaling pathway, as well as the efficacy of Bevacizumab treatment in reducing this effect. The findings revealed that METH exposure enhanced cardiac microvascular permeability while also activating the VEGF-PI3K-Akt-eNOS signaling pathway. Furthermore, treatment with Bevacizumab has been shown to be effective in reversing the METH-induced phenomena. Briefly stated, our research may provide fresh insight into the molecular underpinnings of METH-induced cardiac microvascular permeability, and it may also provide evidence for a relationship between METH misuse and Bevacizumab medication.
Keywords
Introduction
Methamphetamine (METH), a highly addictive and strong amphetamine derivative, is an illegal psychostimulant. 1 At the global level, METH use is a serious public health crisis, with estimated 37 million active users and 2.6 million disability-adjusted life years lost in 2016. 2
Previous research on METH has tended to focus on the drug’s neurotoxicity.3–5 METH, on the other hand, causes numerous organ harm in users.6–8 In recent years, research has revealed that METH damage to the heart was also a key target.9,10 The majority of investigations on METH’s effects on the heart have focused on its histopathological consequences to far.10,11 Daily dosing of rats with METH (3–5 mg/kg, ip) causes histopathological changes in the myocardium, including contraction bands and myocyte degeneration (for 1 week), mitochondrial degeneration with disrupted cristae, myofibrillar hypercontraction, sarcoplasmic reticulum enlargement, and myofilament loss (for 2 weeks). 12 METH usage has been linked to angina, tachycardia, hypertension, myocarditis, dilated cardiomyopathy, arrhythmia, and sudden death in a growing number of clinical and postmortem studies.13,14 However, the processes driving METH-induced cardiac microvascular permeability are still unknown. Fortunately, our previous study showed that METH enhanced the permeability of rats cardiac microvascular endothelial cells (CMECs). Therefore, we speculated that METH could cause cardiac microvascular permeability. Nevertheless, the processes underlying this characteristic have yet to be discovered.
Vascular endothelial growth factor (VEGF) is an important endothelial-specific pro-angiogenic growth factor whose expression is influenced by a variety of variables, including oxygen and metabolite levels. 15 It’s worth noting that VEGF, also known as “vascular permeability factor”, promotes microvascular permeability. 16 Following METH treatment, neurotrophic factors such as VEGF increased expression, according to recent studies.17,18 eNOS, on the other hand, is a cardiovascular guardian angel. In mice, however, when eNOS is knocked out, cardiovascular homeostasis is compromised. 19 Phosphorylation of eNOS appears to be a key regulator of eNOS activity. 20 In addition, PI3K/AKT signaling also controls microvascular permeability and angiogenesis in endothelial cells. 21 However, the consequences of the VEGF-PI3K-Akt-eNOS signaling pathway on METH-induced cardiac microvascular permeability remain unknown. There are a variety of pharmacological drugs that target VEGF receptor signal transduction. One of the most extensively used angiogenic inhibitors, Bevacizumab, is a monoclonal antibody that works directly against VEGF, reducing endothelial cell proliferation and angiogenesis.22–24 But, whether Bevacizumab works against METH-induced cardiac microvascular permeability via the VEGF-PI3K-Akt-eNOS signaling pathway, which is the study’s second issue, needs to be examined further.
The objective of this study was look into the processes of METH-induced cardiac microvascular permeability, with a particular focus on Bevacizumab’s participation in this. Our findings showed that the VEGF-PI3K-Akt-eNOS signaling pathway was important in METH-induced cardiac microvascular permeability, which could be reversed with Bevacizumab treatment. To summarize, our study may offer fresh insight into the molecular mechanisms behind METH-induced cardiac microvascular permeability, as well as new evidence for a relationship between METH misuse and Bevacizumab medication.
Methods
Materials
METH (>99% purity) was obtained from the National Institutes for the Control of Pharmaceutical and Biological Products (Beijing, China). Cell culture reagents, including fetal bovine serum (FBS), DMEM medium and trypsin, were purchased from GIBCO (Carlsbad, CA, USA). ECM medium and Bevacizumab were purchased from Meilunbio (Dalian, China). Peroxidase was purchased from Solarbio, China (Beijing, China). Anti-VEGF, anti-PI3K, anti-phospho Ser473 AKT, and anti-p- eNOS were purchased from Abcam (Cambridge, UK). Anti-AKT (phospho S473), anti-eNOS and anti-GAPDH were purchased from Cell Signaling Technology (Boston, MA, USA). AKT (phospho S473) antibody (ab81283) detects AKT phosphorylated at Serine 473. Other chemicals or reagents, unless specifically described below, were obtained from Sigma-Aldrich (St Louis, MO, USA).
Animal protocol
Adult male C57 BL/6 mice (18–22 g, 6–8 weeks old, purchased from the Laboratory Animal Center of Southern Medical University, Guangzhou, China) were individually housed in tub cages in a temperature-controlled room with a 12 h light-12 h dark cycle and food and water available ad libitum. All animal procedures were carried out according to the NIH Guidelines for the Care and Use of Laboratory Animals and were approved in advance by the Institutional Animal Care and Use Committee at the Southern Medical University. Animals were habituated to the animal facilities for 1 week before treatment. Mice were divided randomly into 3 groups (n = 6/group): Control group, METH group, and Bevacizumab + METH group. METH was dissolved in saline. The Control group received a similar volume of saline. Bevacizumab 5 mg/kg intravenously (i.v.), twice a week for 4 weeks. Mice in the METH exposure group received 2 intraperitoneal (i.p.) injections of METH at 10 mg/kg/injection and 12 h intervals every day for 4 weeks. No animals were excluded in the process. Animals were sacrificed 24 h after the last injection. Heart samples were rapidly removed, then fixed with 4% paraformaldehyde (PFA) and used for double immunofluorescence staining.
Cellculture and sample treatment
Rat primary CMECs were isolated from Sprague Dawley rats (2–4 w, purchased from the Laboratory Animal Center of Southern Medical University). Rats were anesthetized with isoflurane and disinfected with 75% ethanol, and the heart tissue was immediately removed and put into D-Hank’s solution (Procell, China) on ice. After the remaining blood was pumped out, the left ventricle was separated with the epicardial and endocardial cells inactivated by 75% ethanol. The tissue was fully cut and moved into a 50 mL centrifuge tube after 1/4 of the left ventricle was removed, and then 0.2% type II collagenase (Solarbio, China, China) and EDTA-free trypsin (Gibco,USA) were added successively for digestion, and an equal volume of complete medium (composed of ECM, 2.5% FBS, 1% ECGS and 1% penicillin and streptomycin) was added to terminate the digestion. The undigested tissue was filtered with a 70 μm filter, and the cell pellet was collected and inoculated in 60 mm Petri dish coated with 1% gelatin, cultured at 37°C with 5% CO2 for 2 h using ECM complete medium. Finally, the non-adherent cells were removed and cultured with complete medium containing heparin (50 μg/mL, Solarbio, China). The CMECs used in the experiment were the third and fourth generation cells. Cells were plated in six-well plates (5×105 cells/well), incubated at 37°C in a humidified atmosphere containing 5% CO2 throughout the experiments. CMECs were exposed to 0, 0.2, 0.4, 0.6, 0.8 and 1.0 mM METH for 24 h for dose-dependent experiments, and exposed to 0.4 mM METH for 0, 2, 6, 12, 24 and 36 h for time-dependent experiments. Then collected the cells for western blot.
Human umbilical vein endothelial cells (HUVECs) were cultured in DMEM containing 10% FBS, then plated in six-well plates (5×105 cells/well), incubated at 37°C in a humidified atmosphere containing 5% CO2 throughout the experiments. HUVECs were exposed to 0, 0.75, 1.0, 1.25, 1.5 and 2.0 mM METH for 24 h for dose-dependent experiments, and exposed to 1.25 mM METH for 0, 2, 6, 12, 24 and 36 h for time-dependent experiments. Then collected the cells for western blot.
Permeability detection
Horseradish peroxidase assay was used the transwell chamber (Millipore, illerica, USA) to detect the endothelial cell permeability ability of METH. 25 The CMECs or HUVECs were starved for 12 h, and then awaited detection after addition of appropriate medium. After placing an 8 μm transwell chamber in the culture plate, appropriate medium was added. The upper and lower chambers were then separated by a polycarbonate membrane (8 μm). Finally, the cells were inoculated in the upper chamber of matrigel mixed gel (BD Biosciences, USA). After 48 h, the cells were fixed with 4% PFA and then stained with 0.1% crystal violet. Finally, the invasion was observed with a microscope and the number of cell transfers was counted.
siRNA and transient transfection
Specific siRNA sequences targeting VEGF were synthesized by Gene Pharma (Shanghai, China) as follows: 5′-GGCAGCUUGAGUUAAACGATT-3’. CMECs or HUVECs were seeded into 24-well culture plates and transfected with 100 μmol siRNA using Lipofectamine 3000 reagent (Invitrogen, USA), when the cells achieved 30–50% confluent. After 24 h, cells were exposed to 0.4 mM METH for another 24 h. Besides, Bevacizumab 2.0 mM (Meilunbio, China) and 0.4 mM METH were added into cells at 70–90% confluent at the same time for 24 h to serve as the positive control. And then cells were harvest for testing.
Western blot
CMECs and HUVECs exposed to vehicle or METH were lysed in ice-cold RIPA buffer with protease inhibitors and phosphatase inhibitors. Protein concentrations were measured with the BCA-100 Protein Quantitative Analysis kit (Biocolors, Shanghai, China). Protein samples (10 μg) were separated by 6–12% SDS-PAGE and transferred onto PVDF membranes (Millipore, Billerica, MA, USA). The membranes were incubated at room temperature for 2 h in blocking buffer (5% BSA or 5% nonfat dry milk in TBST buffer), followed by incubation with primary antibodies [anti-VEGF, anti-PI3K, anti-AKT, anti-phospho Ser473 AKT, anti-eNOS and anti-p-eNOS, anti-GAPDH (1:1000 for all)] overnight at 4°C. After washing the membranes three times with TBST, they were incubated with an anti-rabbit IgG-horseradish-peroxidase (1:10,000, Jackson, MI, USA) for 1h at room temperature. The membranes were developed with ECL detection reagents and the signal of band intensities was quantitated with Gel-Pro analyzer. Expression of the housekeeping gene GAPDH was used as a reference control. Western blot experiments for each protein of interest were repeated three times. Only one representative band for each protein is shown in this manuscript.
Fluorescence microscopy
The paraffin sections of heart were dewaxed, and the antigen was repaired using a heating method. The slices were blocked using 10% goat serum for 1 h, and incubated with primary antibody overnight at 4°C. The following steps were carried out in the dark. The sections were incubated at 37°C for 1 h with fluorescent labelled secondary antibody of appropriate concentration. DAPI was dripped and observed under fluorescence microscope.
Statistical analysis
Data are summarized as mean ± SEM of at least 3 independent replicates. Parametric and nonparametric tests (as appropriate) were used to analyze the data using SPSS 20.0 software. The parametric test was one-way ANOVA, followed by LSD post hoc test. The nonparametric test included Mann-Whitney U in two-independent sample test or Kruskal-Wallis H in K independent samples test and the post hoc test was Bonferroni method. p < 0.05 was considered statistically significant.
Results
METH exposure increased endothelial cell permeability and VEGF expression
We employed a transwell experiment in HUVECs and CMECs to demonstrate METH’s potential to increase endothelial cell permeability. Both studies showed that METH causes an increase in endothelial cell permeability (Figure 1(a–d). Next, we measured VEGF protein expression in HUVECs treated with a dosage range of (0–2.0 mM) METH for 36 h. When 1.50 mM METH was administered, VEGF clearly reached its maximal protein concentration after 24 h Figure 1(e). And the concentration of VEGF start increased at 2 h after administration, as seen in Figure 1(f). Finally, using a dose range of 0–1.0 mM of METH for 36 h, we investigated the involvement of VEGF in METH-induced CMECs. Up to the maximum concentration tested at 24 h, METH concentrations of 0.40 mM resulted in a significant increase in VEGF protein release, which gradually declines (Figure 1(g and h)). METH exposure enhances endothelial cell permeability and VEGF expression, according to these findings. We used METH 1.50 mM (HUVECs) and 0.40 mM (CMECs) as the concentration for the next tests based on the previous results. The best condition was found to be 24 h of METH incubation. METH exposure increased VEGF protein expression in HUVEC and CMECs cells. (a–b) Detection of filtered factor content in a transwell chamber using horseradish peroxidase assay in HUVECs. After 1.5 mM METH treatment for 24 h, the absorbance was the highest. (c–d) In the transwell test of the CMECs, after 0.4 mM METH treatment for 24 h, the absorbance was the highest. (e–f) The expression of VEGF increased most significantly after 1.5 mM METH treatment for 24 h in the HUVECs. (g–h) The expression of VEGF increased most significantly after 0.4 mM METH treatment for 24 h in the CMECs. The data were expressed as mean ± SEM (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001 vs. Control group.
siVEGF knockdown in HUVECs and CMECs reduced METH-induced endothelial hyperpermeability
The effects of METH on VEGF expression on endothelial hyperpermeability in HUVECs and CMECs, i.e. the primary signaling route downstream of METH stimulation, were then investigated.
To begin, HUVECs were treated with METH (1.50 mM) for 24 h, and protein expression were evaluated using western blot. METH administration increased the expression of VEGF, PI3K, phospho Ser473 AKT, and p-eNOS proteins (Figure 2(a and b)). However, when compared to the METH treatment group, siRNA dramatically lowered these expression (Figure 2(a and b)). Then, CMECs were cultured in full medium with METH (1.50 mM) for 24 h. While METH alone enhanced the expression of VEGF, PI3K, phospho Ser473 AKT, and p-eNOS proteins, when cells were treated with siRNA+METH, the above mentioned proteins remained unchanged or even reduced (Figure 2(c–d)). In HUVECs and CMECs, however, there was no difference in AKT and eNOS expression with or without METH or siRNA treatment (Figure 2). Inhibition of VEGF reduced METH-induced P13 K, phospho Ser473 AKT, and p-eNOS activation in HUVECs and CMECs. HUVECs (a–b) and CMECs (c–d) were transfected with si-NC or si-VEGF for 48 h, and then treated with or without 1.5 mM METH, 0.4 mM METH for 24 h. Western Blot and quantitative analyses were performed to determine the expression of VEGF/PI3K, phospho Ser473 AKT/AKT (phospho S473), peNOS/eNOS proteins. The data were expressed as mean ± SEM (n = 3). **p < 0.01, and ***p < 0.001 vs. si-NC group. #p < 0.05, ##p < 0.01, and ###p < 0.001 vs. si-NC+METH group.
Bevacizumab decreased METH-induced endothelial hyperpermeability through inhibiting the VEGF-PI3K-Akt-eNOS signaling
For further verification, Bevacizumab was performed. We used Bevacizumab to inhibit the endothelial hyperpermeability of HUVECs and CMECs induced by the administration of METH. In HUVECs, the analysis showed decreased permeability in the BEVA+METH group, compared with the METH group (Figure 3(a)). Moreover, Bevacizumab significantly antagonized the effects of METH. That is, Bevacizumab not only prevented VEGF, but also decreased PI3K, phospho Ser473 AKT, and p-eNOS protein expression in the HUVECs (Figure 3(b and c)). Nevertheless, there were no differences in AKT and eNOS protein expression (Figure 3(b and c)). The same results were seen in CMECs (Figure 3(d–f)). Bevacizumab inhibited METH-induced VEGF-PI3K-Akt-eNOS signaling pathway. Bevacizumab treatment inhibited HUVECs mobility induced by METH (a), and reduced the expression levels of VEGF, P13 K, p-AKT, p-eNOS (b–c). Likewise, when treatment with Bevacizumab, the CMECs mobility with METHinduced (d) and expression levels of VEGF, P13 K, p-AKT, p-eNOS (e–f) were reduced. The data were expressed as mean ± SEM (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001 vs. Control group; #p < 0.05, ##p < 0.01, and ###p < 0.001 vs. Bevacizumab + METH group.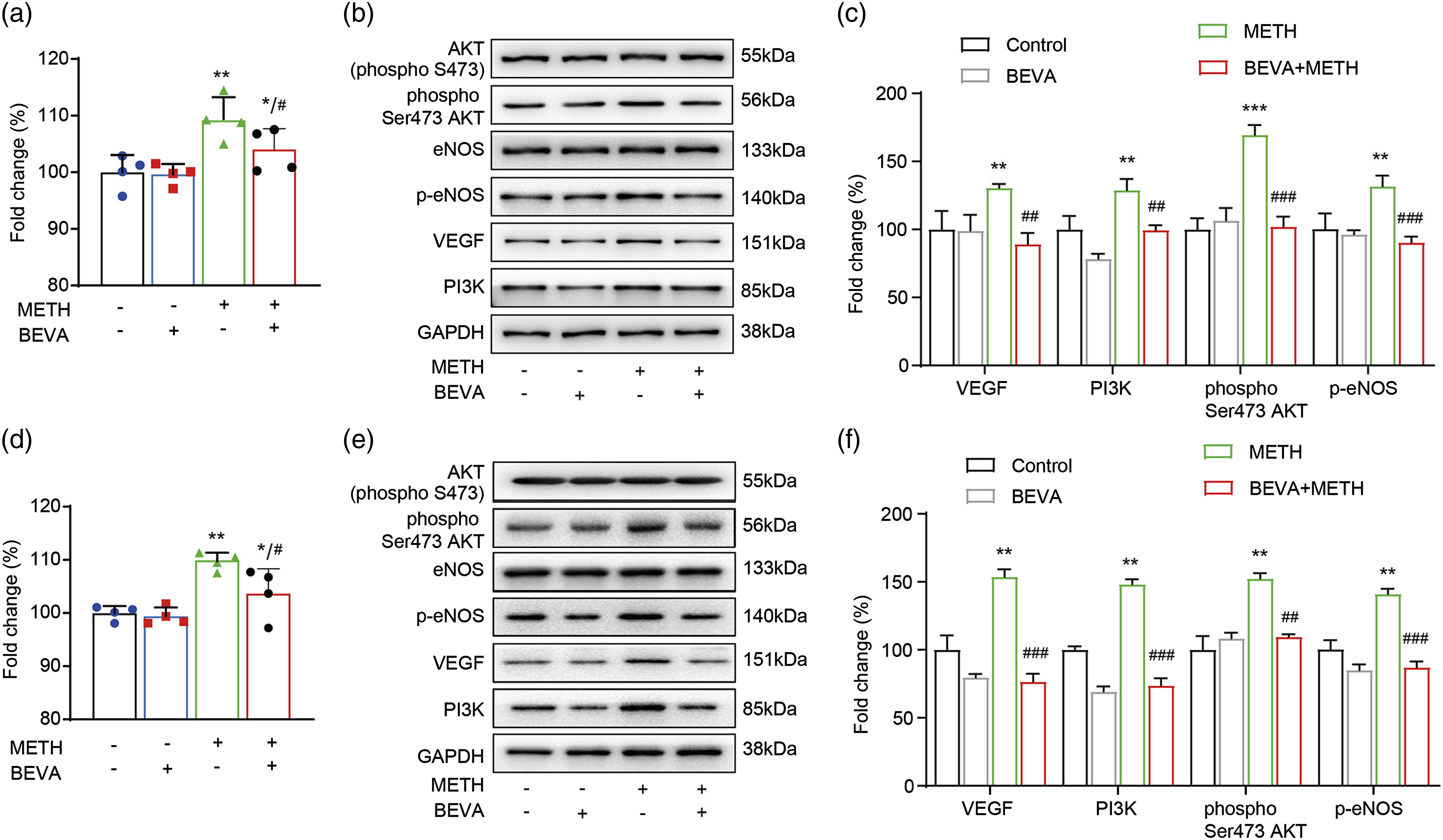
Bevacizumab reversed METH-induced cardiac microvascular permeability in mice by suppressing VEGF-PI3K-AKT-eNOS signaling
Lastly, to determine the METH-induced cardiac microvascular permeability in vivo, we performed animal experiments. To generate the endothelial cell hyperpermeability mouse model, mice were chemically induced by METH. Firstly, by comparing the Control group, we can see that METH significantly increased VEGF expression (green fluorescence, Figure 4). And then, the expression of PI3K (red, Figure 4(a)), phospho Ser473 AKT (red, Figure 4(b)), and p-eNOS (red, Figure 4(c)) were also upregulated. However, those phenomena were reversed with the addition of Bevacizumab. The results shown in Figure 4 indicate that Bevacizumab significantly downregulated the VEGF, PI3K, phospho Ser473 AKT, and p-eNOS protein fluorescence intensity. Inhibition of permeability markers by Bevacizumab reverses meth-induced increases in permeability. The expressions of VEGF/PI3K (a), VEGF/phospho Ser473 AKT (b), and VEGF/p-eNOS (c) in the METH group were significantly up-regulated compared to the control group, but the Bevacizumab was able to dramatically down-regulate the expressions of them. n = 6 mice per group. Scale bar = 50 μm.
Discussion
METH is an extremely dangerous substance, widely regarded as the most poisonous and addictive, 26 because it stimulates 10 times the normal amount of dopamine production and causes considerable harm. 27 METH induces cardiac injury by multifactorial pathways, according to a growing number of studies published in the previous few years. 28 METH-associated cardiomyopathy is more common in long-term METH users, 29 but it’s possible that a large dose of METH caused acute damage to a heart that was previously damaged. 30 However, METH has not been linked to cardiac microvascular permeability so far.
Our data show that METH exposure increases endothelial cell permeability. Transwell assays revealed that METH increased permeability across HUVECs and CMECs (Figure 1(a–d)). In addition, METH-treated cells have higher VEGF expression. 31 Our findings backed up this assertion (Figure 1(e–h)). Further research was carried out as a result of this. METH boosted the expression of VEGF and PI3K in HUVECs and CMECs, as well as the phosphorylation of AKT and e-NOS, according to our findings (Figure 2). In other words, METH triggered the VEGF-PI3K-Akt-eNOS signaling pathway. This pathway was also blocked after silencing VEGF using siRNA. As a result, we believe that METH activated the VEGF-PI3K-Akt-eNOS signaling cascade, increasing endothelial cell permeability. The VEGF inhibitor, on the other hand, completely restored the aforementioned alterations. VEGF inhibitors, particularly Bevacizumab, have long been recognized to bind circulating VEGF and prevent it from attaching to its receptor.32,33 In fact, there are three similar VEGF inhibitors, namely VEGF Trap, Ranibizumab and Bevacizumab. First, VEGF Trap has a high affinity for VEGF-A and PlGF isoforms in all mammalian species, whereas both Ranibizumab and Bevacizumab bind and neutralize mouse or rat VEGF-A with relatively low affinity. Second, Ranibizumab was 30-100-fold more potent than the monovalent Fab fragment of Bevacizumab in bioassays measuring VEGF-induced endothelial cell mitosis. In conclusion, Bevacizumab is the three mAbs with the lowest affinity for the VEGF-A target in mammals. However, the VEGF-PI3K-Akt-eNOS signaling pathway triggered by METH was blocked with the least and mildest Bevacizumab. Furthermore, it is inferred that high-affinity inhibition of the target VEGF-A can effectively counteract the METH-induced increases endothelial cell permeability. As shown in Figure 3, VEGF, PI3K, phospho Ser473 AKT and p-eNOS levels decreased significantly at 24 h after Bevacizumab treatment, compared to METH group. Bevacizumab down-regulated above mentioned proteins expression possibly through the direct effect of it on VEGF. Therefore, it is speculated that Bevacizumab inhibited VEGF and might block the VEGF-PI3K-Akt-eNOS signaling cascade, reducing METH-induced increased permeability in endothelial cells. Finally, the current study’s in vivo trials corroborated this. METH enhances cardiac microvascular permeability in mice through stimulating the VEGF-PI3K-Akt-eNOS signaling cascade, while Bevacizumab reverses this effect (Figure 4).
METH enhances cardiac microvascular permeability through activating the VEGF-PI3K-Akt-eNOS signaling cascade, which is blocked by a VEGF inhibitor (particularly Bevacizumab). Our findings are in line with postmortem findings of myocardial bleeding. This is the first time we’ve heard about it. Increased cardiac microvascular permeability is one significant future route of METH-induced harm. Alternatively, VEGF inhibitors or Bevacizumab may be effective in treating METH-induced heart microvascular permeability.
These investigations have made some important discoveries, but they also have certain drawbacks. On the one hand, there were missing data on electron microscope images in our investigation. The absence of cardiac pathology in mice, on the other hand, constituted a challenge. Therefore, we’ll continue to investigate this issue in the next experiment.
Conclusions
Our study provides new insight into the molecular mechanisms of METH-induced cardiac microvascular permeability and new evidence for the treatment with VEGF inhibitors following METH abuse.
Footnotes
Acknowledgements
Special thanks to the Laboratory of department of Forensic Medicine Southern Medical University for the help of this project.
Authors’ contributions
HJW and JPT conceived, designed and coordinated the study. RC performed the in-vitro. PH completed the writing of manuscript and figures. SRW performed the vivo studies. CZ collected and assembled data. XL analyzed and interpreted data. All authors read, and approved the submitted version.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
This study is approved by the ethical committee of Southern Medical University (L2020064).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for these studies was provided by the National Natural Science Foundation of China (81901362); the Natural Science Foundation of Guangdong Province (2018A030310122); the Doctor Initiation Fund of Guangdong Medical University (B2017029); the Bureau of Science and Technology of Foshan City Project (1920001000475, and 2020001005703); and Innovation Project of Women and Children medical research center affiliated to Foshan Institute of Fetal Medicine (FEYJZX-2021-003 and FEYJZX-2021-004).