
Abstract
Mitochondrial dysfunction was considered to be a critical event in acetaminophen (APAP) -induced hepatotoxicity. Recent studies suggest that abnormal mitochondrial dynamics contributes to mitochondrial dysfunction in APAP-induced liver injury, yet the underlying mechanisms responsible for deregulated mitochondrial dynamics remains elusive. In this study, C57BL/6 mice were used to establish a model of acute liver injury via intraperitoneal (i.p.) injection with overdose of APAP. Furthermore, calpain intervention experiments were achieved by the inhibitors ALLN or calpeptin. The activity of serum enzymes and pathological changes of APAP-treated mice were evaluated, and the critical molecules in mitochondrial dynamics and calpain degradative pathway were determined by electron microscopy, immunoblot and calpain activity kit. The results demonstrated that APAP overdose resulted in a severe liver injury, mitochondrial damage and an obvious cleavage of fusion/fission proteins. Meanwhile, the activation of calpain degradative machinery in liver were observed following APAP. By contrast, pretreatment of calpain inhibitors significantly inhibited the activation of calpains. Our further investigation found that ALLN or calpeptin administration significantly suppresses the changes of mitochondrial dynamics in APAP-treated mice and finally protected against APAP-induced hepatoxicity. Overall, these results suggest that calpain-mediated cleavage of mitochondrial dynamics proteins was involved in the pathogenic process of mitochondrial dysfunction and thus present a potential molecular coupling APAP-induced hepatotoxicity.
Introduction
Acetaminophen is the most widely used analgesic/antipyretic drug in the world. Although APAP is commonly considered to be efficacious and safe at therapeutic doses, a high dose of APAP can result in severe liver injury, which is responsible for nearly half of acute liver failure in the United States and many European countries.1,2 APAP hepatotoxicity is characterized by massive hepatocyte damage primarily in the centrilobular region of the liver. The metabolic activation of APAP is generally considered to be obligatory for APAP hepatotoxicity. In particular, the electrophilic metabolites such as N-acetyl-p-benzoquinone imine (NAPQI) generated from APAP metabolism are capable of covalently modifying cellular macromolecules.
In the past decades, much research has been done to implicate mitochondrial dysfunction as an early prominent feature in pathogenic mechanism of APAP-induced liver injury. Such a primary role is underscored by the fact that NAPQI binds mitochondrial membrane proteins to subsequently initiate mitochondrial membrane permeabilization and oxidant stress, resulting in collapse of the membrane potential and consequently cell death.3,4 Complex I and III are refractory to APAP-mediated disruption in liver mitochondria, but APAP disrupts their formation in hepatocytes in vitro and in vivo, which leads to increased production of reactive oxygen species (ROS). 5 However, the exact mechanisms underlying mitochondrial dysfunction in APAP-induced hepatotoxicity remain incompletely understood.
Recent studies from multiple groups demonstrated that abnormal mitochondrial dynamics were causally involved in APAP‐induced mitochondrial dysfunction. For instance, 66Shc, a major regulator of mitochondrial ROS, played an essential role in mediating mitochondrial dynamics perturbation and mitochondrial fragmentation in response to APAP, both in vivo and in vitro. 6 Furthermore, in APAP-induced liver injury, p53 up-regulated modulator of apoptosis (PUMA) was accumulated in mitochondria, and PUMA KO suppressed APAP-induced mitochondrial dynamics imbalance. 7 Mitochondrial numbers were reduced and became scattered and fragmented, exhibiting short rods or sphere shapes in mice treated with APAP revealed by transmission electron microscopic (TEM) imaging study. 6 APAP hepatotoxicity is an intricate process that involves mitochondrial oxidant stress, mitochondrial fragmentation, and hepatocyte cell death. 7
Mitochondria are highly dynamic organelles that undergo continuous fission/fusion events that play a critical role in maintaining mitochondrial homeostasis. These processes are tightly regulated by several principal proteins; for example, mitochondrial fusion is controlled by mitofusin1(Mfn1), mitofusin2 (Mfn2), and optic atrophy 1 (Opa1), whereas mitochondrial fission is controlled by dynamin-related protein 1(Drp1) and mitochondrial fission 1 (Fis1). 8 Unopposed fission leads to fragmentation whereas unopposed fusion causes elongation, both of which could impair mitochondrial function.9,10 Thus, not surprisingly, emerging studies suggest the altered balance in mitochondrial fission and fusion might be a potential mechanism leading to mitochondrial dysfunction in multiple diseases.
There is increasing evidence supporting that intracellular Ca2+ homeostasis is disrupted in APAP hepatotoxicity, which affects diverse signaling cascades linked to the mitochondrial dysfunction and hepatocyte death. For example, a sustained elevation of the free cytosolic Ca2+ concentration in hepatocytes was observed, which was believed to be a secondary event of oxidative stress following APAP.11,12 Mechanistically, covalent binding of the reactive metabolite of APAP to the calcium ATPase might be responsible for the loss of its activity.11,13 As the calcium-dependent neutral cysteine proteases, calpains are widely involved in the maintenance of cell survival and death.14–19 Importantly, recent research found that activated calpain can degrade Drp1, MFN1/2 and other regulatory proteins in the neurodegenerative diseases,20,21 which supported a mechanistic link between calpain and mitochondrial dynamics. Given the similarity between dynamin and dynamin‐related proteins involved in mitochondrial fission and fusion, we hypothesized that calpain activation may be involved in the changes of these mitochondrial fission/fusion proteins in APAP-induced hepatotoxicity.
In the present study, we aimed to investigate the proteolysis and posttranscriptional modifications of mitochondrial fission/fusion GTPases with a focus on the effects of calpain activation. We have found that APAP overdose leads to the change of mitochondrial dynamics and the activation of calpain in mice livers. Inhibition of calpain by N-Acetyl-Leu-Leu-norleucinal (ALLN) or calpeptin significantly decreased the cleavage of MFn2 and Drp1and prevented APAP-induced acute liver injury. Our work therefore highlights that calpain plays a primary role in cleavage of mitochondrial fusion/fission proteins in acetaminophen-induced mice liver injury.
Materials and methods
Materials
APAP (purity > 99%) was purchased from Sigma-Aldrich Chemical Co (St Louis, MO, USA). N-Acetyl-Leu-Leu-norleucinal(ALLN, purity > 98.3%) was purchased from Calbiochem/EMD Chemicals Inc (San Diego, CA, USA). Calpeptin(purity ≥ 98.0%)was purchased from Med Chem Express (Monmouth Junction, NJ, USA). The kits for analysis of alanine/aspartate were obtained from Bioengineering Institute (Beijing, China). Primary antibodies used in this study include anti-calcineurin (BD biosciences, #610259), μ-Calpain (Sigma-Aldrich, #C0355), m-Calpain (Sigma-Aldrich, #C268), Calpastatin (Sigma-Aldrich, #C270), Drp1/Dlp1(BD biosciences, #611112), p-Drp1(Ser616) (CST, #3455s), p-Drp1(Ser637) (CST, #4867s), MFn2(Santa Cruz, #sc-100560). Monoclonal anti-β-actin, peroxidase-Conjugated Goat anti-Rabbit and Goat anti-Mouse IgG secondary antibodies, and Protease Inhibitor Cocktail were purchased from Sigma-Aldrich Chemical Co. GENMED Calpain Activity Assay Kits are from GENMED Scientifics inc. (Arlington, MA, USA). Mitochondria Isolation Kit are from Beyotime Biotechnology, China. NE-PER Nuclear and Cytoplasmic Extraction Reagents Kits, BCA Protein assay Kit and SuperSignal® West Pico Chemiluminescent Substrate Kit were purchased from Pierce Biotechnology, Inc. (Rockford, IL, USA). All other chemicals were of analytical grade from commercial supplier.
Animals and treatments
Male C57BL/6 mice were purchased from the Experimental Animal Center of Shandong University. Forty male C57BL/6 mice were randomly divided into four groups to obtain the dose-response model (n = 10 mice/group): (1) control group, (2) 180 mg/kg APAP group, (3) 300 mg/kg APAP group, (4) 500 mg/kg APAP group. After fasting 12 h, mice were intraperitoneally injected with saline or APAP solution and then sacrificed at time-point of 24h. Furthermore, another forty mice were divided into four groups for ALLN intervention experiment (n=10 mice/group): (1) control group, (2) 20 mg/kg ALLN group, (3) 300 mg/kg APAP group, (4) 300 mg/kg APAP + 20 mg/kg ALLN group; forty mice were divided into four groups for Calpeptin intervention experiment (n=10 mice/group): (1) control group, (2) 2 mg/kg Calpeptin group, (3) 300 mg/kg APAP group, (4) 300 mg/kg APAP + 2 mg/kg Calpeptin group. After fasting 12h, mice were intraperitoneally injected with saline or APAP solution. ALLN (20 mg/kg) or Calpeptin (2 mg/kg) was given intraperitoneally to animals of intervention groups 1 h prior to APAP administration. Then, all animals were sacrificed at time-point of 24h. Serum and liver tissue of all groups were collected for further analysis. All experiments were carried out in accordance with the NIH Guide for Care and Use of Laboratory Animals and followed the principles in the “Use of Animals in Toxicology”. All protocols were approved by the Institutional Animal Care and Use Committee of Shandong University.
Serum analyses and histological examination
The blood samples were kept at room temperature for 1h. After centrifugation at 3000 g for 15 minutes, serum was collected to measure the levels of ALT and AST with kits according to the manufacturer’s instructions. Furthermore, a segment of liver was fixed in 4% neutral buffered formalin solution at least 24 h. Then the liver tissues were dehydrated with a sequence of ethanol solutions, embedded in paraffin wax and sectioned at 4 μm thickness. At last, tissue sections were stained with hematoxylin and eosin (HE), and then observed to evaluate histopathological changes under light microscopy.
Transmission electron microscopy analyses of mice livers
Liver samples of control and treated animals were removed and fixed by immersion in ice-cold 3% glutaraldehyde for 2 h. Afterwards, samples were postfixed in OsO4 (1%, 1 h), dehydrated through ethanol series, and embedded in epon resin. Ultra-thin sections (60 nm) were double stained with uranyl acetate and lead citrate, examined by an electron microscope (Hitachi H800).
Calpain activity assay
Calpain activity was assayed using a GENMED Calpain Activity Assay Kit, according to the manufacturer’s instructions. Liver tissues collected from ALLN-intervention experiment mice were homogenized on ice, homogenates were centrifuged at 10,000 rpm for 10 min. After determination of the protein content using the BCA protein assay, aliquots of the supernatants were incubated with the fluorogenic substrate. The assay measures the ability of calpains to digest the synthetic substrate suc-LLVY-AMC. Released the fluorophore (AMC) was measured fluorometrically at an excitation of 380 nm and an emission of 460 nm. Calpains activity was expressed as relative fluorescence units (RFU)/ug protein.
Tissue preparation and immunoblot analysis
Total proteins were extracted from mouse liver using RIPA lysis buffer. Tissue homogenates were centrifuged at 12,000 g for 15 minutes, and then the supernatants were kept for western analysis of target protein. Mitochondrial proteins were extracted from mice livers using the Tissue Mitochondria Isolation Kit. Equivalent amounts of protein samples were separated by SDS-PAGE electrophoresis and transferred to PVDF membranes. Then the membranes were blocked in non-fat milk for 1h at room temperature, incubated with primary antibody overnight at 4°C and incubated with horseradish peroxidase-conjugated secondary antibody at the room temperature. Subsequently, the membranes were detected by chemiluminescence kit. Finally, the immunoreactive bands of proteins were scanned with Agfa Duoscan T1200 scanner, and the digitize data were quantified as integrated optical density (IOD) using Image-Pro Plus software.
Statistical analysis
All data were expressed as the mean±SD. Multiple statistical comparisons between APAP groups and control groups were performed by one-way analysis of variance (ANOVA) using SPSS software, version 20.0. p value less than 0.05 was considered statistically significant.
Results
APAP induced a severe liver injury in mice
APAP overdose caused severe liver injury in mice, as indicated by the increased serum ALT and AST activities and extensive centrilobular necrosis. As shown in Figure 1, a dose-dependent increase in levels of ALT and AST was observed with APAP treatment, which reached the maximum levels in 500 mg/kg APAP group (p < .05). Furthermore, histopathological examination of liver sections showed that liver sections from the APAP groups exhibited various degrees of centrilobular hepatocyte necrosis accompanied with granular degeneration of hepatocytes and infiltration of inflammatory cells. Taken together, our results support that APAP exposure results in a severe live injury in mice. Biochemical assessment and histopathological analysis of mice following APAP treatment. (A) Histopathological analysis of representative HE stained liver samples of APAP-treated mice (dashed line shows necrotic area). Scale bar = 100 μm. (B) Serum ALT and AST activities in APAP-treated mice.
APAP administration resulted in abnormal mitochondrial dynamics and calpain activation in mice
TEM analysis demonstrated some markedly mitochondrial abnormalities such as loss of cristae membranes and swollen and reduced matrix density in the mice liver after APAP treatment. Importantly, shortened mitochondrial length (red double arrows) and decreased size of mitochondria were also observed because of excessive fission in APAP-treated animals compared with the control (Figure 2(a)). Consistent with the morphological changes, APAP exposure caused a significant increase in the level of Drp1. Furthermore, we found that levels of the full-length of MFn2 (∼86 kDa) were decreased and levels of the full-length of Drp1 were increased, but the cleavage of Drp1 and MFn2 was significantly increased in mice liver after APAP treatment. Meanwhile, dephosphorylation of Drp1 at Ser637 site and phosphorylation of Drp1 at Ser616 site were induced by APAP administration. APAP administration causes excessive mitochondrial dynamics deregulation in mice. (A) Electron micrographs of ultrathin sections of mice livers in APAP-treatment mice. The disappearance of cristae membranes and decreased size of mitochondria in mice liver were observed after the treatment of APAP. Red double arrows show differences in mitochondrial length between groups. Scar bar=1 μm. (B) Immunoblot analysis for the critical molecules regulating mitochondria fission in mitochondrial fraction of APAP treatment mice liver. Native Drp1 was demonstrated as about 55 kDa band, cleaved Drp1 was shown in about 25 kDa. (C) Immunoblot analysis for the critical molecule regulating mitochondria fusion in mitochondrial fraction of APAP treatment mice liver. Native MFn2 was demonstrated as about 86 kDa band, cleaved MFn2 was shown to range from about 25 to 55 kDa. The data of target proteins in the experiment groups were normalized to VDAC, and then expressed as the mean percentage of the control (100%), n = 6. The data are representative of three independent experiments. *p < .05 compared to the control group, #p < .05 compared to APAP group.
More importantly, APAP treatment resulted in an activation of calpain degradation pathway. As shown in Figure 3(a), the levels of major components of calpain system, including μ-calpain, m-calpain, and the cleaved products of calpastatin were significantly elevated in a dose-dependent manner. To testify the change of calpain activity in liver tissue, we measured proteolytic activities of calpains in mice liver extracts by using calpain activity kit, and calpain activity in mice treated with APAP was increased in a dose-dependent manner. These results supported that calpain degradative machinery in mice livers was obviously activated following APAP. The changes of calpain degradative machinery in livers of mice treated with APAP. (A) Immunoblot analysis for m-calpain, μ-calpain, and calpastatin in mice liver lysates. The data of target proteins in the experiment groups were normalized to β-actin, then expressed as the mean percentage of the control (100%), n = 6. The data are representative of three independent experiments. (B) The changes of calpain activity in liver tissues of mice. Calpain activity in liver samples was measured using a fluorimetric assay. The data shown are means ± SD (n = 6 samples/group, each performed in duplicate). *p<0.05 compared to the control group.
Calpain inhibitors suppressed the activation of calpain and alleviated APAP-induced the change of mitochondrial dynamics in mice
It has been reported that imbalanced mitochondria dynamic is associated with calpain. 20 To determine the possible role of calpain in APAP-induced the change of mitochondria dynamic, we then treated mice with calpain inhibitors ALLN and Calpeptin prior to APAP administration and measured their effects.
We first testified the effect of ALLN on calpain activity in mice liver, and measured proteolytic activities of calpains in mice liver extracts. As shown in Figure 4(a), calpain activity in mice treated with APAP was increased by 105.7% (p < .05) compared to the control. By contrast, ALLN pretreatment significantly inhibited the increase in calpain activity in the liver of APAP-treated mice. Furthermore, ALLN obviously affected APAP-induced changes in key molecules of calpain degradative pathway. When compared to APAP-treated group, the expression of m-calpain and μ-calpain and the cleavage of calpastatin were also significantly decreased by ALLN pretreatment (Figure 4(b)). These results were consistent with the change of calpain activity in liver tissues of ALLN-pretreated mice. Inhibition of calpain activity in mice liver of intervention experiments with ALLN. (A) The changes of calpain activity in liver tissues of mice. Calpain activity in liver samples was measured using a fluorimetric assay. The data shown are means ± SD (n = 6 samples/group, each performed in duplicate). *p < 0.05 compared to the control group, #p < 0.05 compared to APAP group. (B) Immunoblot analysis for m-calpain, μ-calpain, and calpastatin in mice liver lysates. The data of target proteins in the experiment groups were normalized to β-actin, then expressed as the mean percentage of the control (100%), n = 6. The data are representative of three independent experiments. *p < 0.05 compared to the control group.
Then, the change of mitochondrial damage was further confirmed. First, mitochondrial morphology was analyzed through TEM (Figure 5(a)). We observed that mitochondrial ultrastructure was obviously damaged in APAP group. By contrast, ALLN pretreatment significantly alleviated mitochondrial structure damage, for example cristae membranes, matrix density and the size of mitochondria all markedly reduced to the margin level of normal mice after APAP treatment. Importantly, further analysis of TEM results showed that mitochondria in liver treated with APAP displayed a fragmented morphology compared with control group, as evidenced by a decrease in aspect ratio, higher circularity and a significant shift towards shortened length (< 0.5 μm) (Figure 5(b)). The total number of mitochondria dropped greatly upon APAP insults, indicating a reduction of mitochondrial biomass. Compared with APAP group, ALLN intervention significantly restored mitochondrial dynamics. Next, we have examined the changes in mitochondria dynamics proteins. As showed in Figure 5(c), ALLN pretreatment caused a significant decrease in the level of Drp1 and an increase in the level of MFn2. More importantly, the cleavage of Drp1 and MFn2 was significantly decreased in mice liver by ALLN pretreatment. These data support that ALLN inhibits mitochondrial excessive fission in APAP-induced acute liver injury model. The changes in mitochondria dynamics pathway in mice liver with ALLN and calpeptin pretreatment. (A) Electron micrographs of ultrathin sections of mice livers in ALLN pretreatment mice. The disappearance of cristae membranes and decreased size of mitochondria in mice liver were alleviated by ALLN after the treatment of APAP. Red double arrows show differences in mitochondrial length between groups. Scar bar=1 μm. (B) Quantification of mitochondrial morphological parameters and number. The boundaries of 25th and 75th percentiles are plotted in graphs for mitochondria aspect ratio, circularity (n = 200 mitochondria per group) and number per micrograph (n = 10 per group). Mitochondria are classified into three different groups according to their length (<0.5 μm, 0.5–1.0 μm and >1.0 μm) and their relative quantities were calculated (n = 200 mitochondria per group). (C) Immunoblot analysis for the critical molecules regulating mitochondria dynamics in mitochondrial fraction of ALLN pretreatment mice liver. (D) Immunoblot analysis for calcineurin and p-Drp1 in ALLN pretreatment mice liver lysates. (E) Immunoblot analysis for the critical molecules regulating mitochondria dynamics in mitochondrial fraction of calpeptin pretreatment mice liver. The data of target proteins in the experiment groups were normalized to β-actin or VDAC, and then expressed as the mean percentage of the control (100%), n = 6. The data are representative of three independent experiments. *p < .05 compared to the control group, #p < .05 compared to APAP group.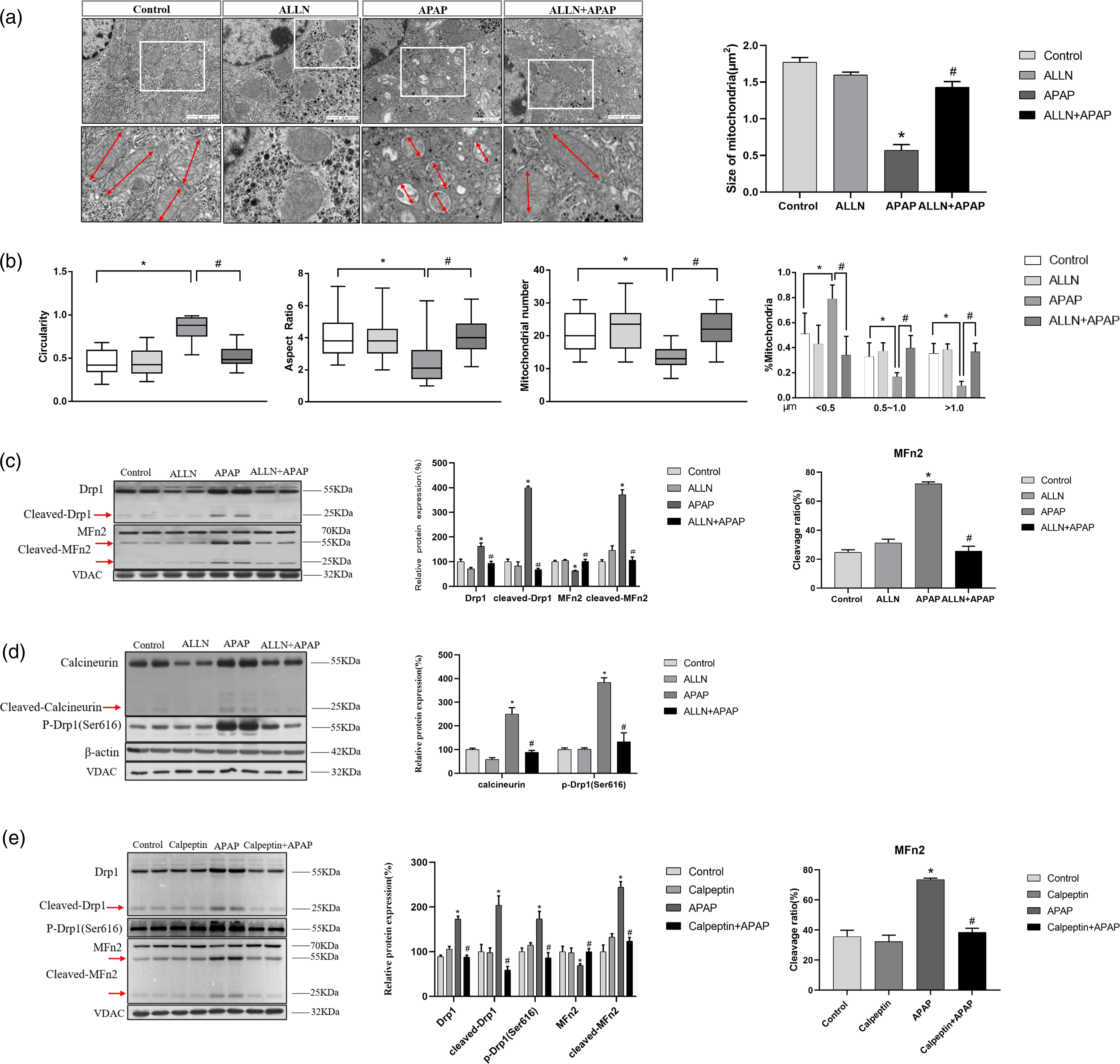
Calcineurin is a known Ca2+/calmodulin-dependent serine/threonine phosphatase. 22 And mitochondrial fragmentation and Drp1 phosphorylation at Ser637 are regulated by calcineurin through their phosphatase activity. 23 The levels of cleaved calcineurin were decreased in mice liver with ALLN pretreatment compared with that only APAP treatment. Our results also showed that the increased level of phosphorylation at Drp1 (Ser 616) was reduced by pretreatment with ALLN. These results suggested that calpain signals are key regulators in APAP-induced mitochondrial fragmentation and liver cell death.
To further identify the effects of calpain degradative pathway on APAP-induced liver injury, C57BL/6 mice were treated with 2 mg/kg calpeptin (a more specific inhibitor for calpain) prior to APAP administration. As expected, pretreatment with calpeptin further confirmed that the inhibition of calpain prevented mitochondrial fragmentation. Immunoblot analysis revealed that calpeptin resulted in a lower level of the full length of Drp1 and a lower expression of the cleavage of Drp1 in the mice liver compared to the APAP-treated group. In the meantime, calpeptin pretreatment markedly suppressed the elevation of the level of phosphorylation at Drp1 (Ser616) induced by APAP (Figure 5(e)). Furthermore, we examined the expression of mitochondrial fusion protein in mice livers. MFn2, but not other mitochondrial fusion proteins such as MFn1 and OPA1, was decreased by APAP that was effectively prevented by calpeptin. Interestingly, significantly fewer cleavage fragments of MFn2 were observed by calpeptin compared with APAP treatment. Not surprisingly, the calpain inhibitor calpeptin resulted in significant inhibition of mitochondrial fragmentation.
Calpain inhibitors pretreatment reduced APAP-induced acute liver injury
Pretreatment with ALLN significantly attenuated the increase in ALT and AST activities and decreased areas of liver necrosis in APAP-treated mice (Figure 6). In particular, the activity of serum ALT and AST in mice treated with 20 mg/kg ALLN prior to APAP was markedly reduced to the margin level of normal mice, although a significant difference was still observed when compared with the control. Furthermore, histopathological analysis further confirmed the biochemical assessment of the ALLN intervention experiment. Biochemical assessment, histopathological analysis of mice in intervention experiments with ALLN and Calpeptin. (A) Liver histopathological analysis of ALLN intervention experiment. Histopathological analysis of representative HE stained liver samples of each group in intervention experiment (dashed line shows necrotic area). Scale bar = 100 μm. (B) Serum ALT and AST activities in ALLN intervention experiment. (C) Liver histopathological analysis of calpeptin intervention experiment. Histopathological analysis of representative HE stained liver samples of each group in intervention experiment (dashed line shows necrotic area). Scale bar = 100 μm. (D) Serum ALT and AST activities in calpeptin intervention experiment.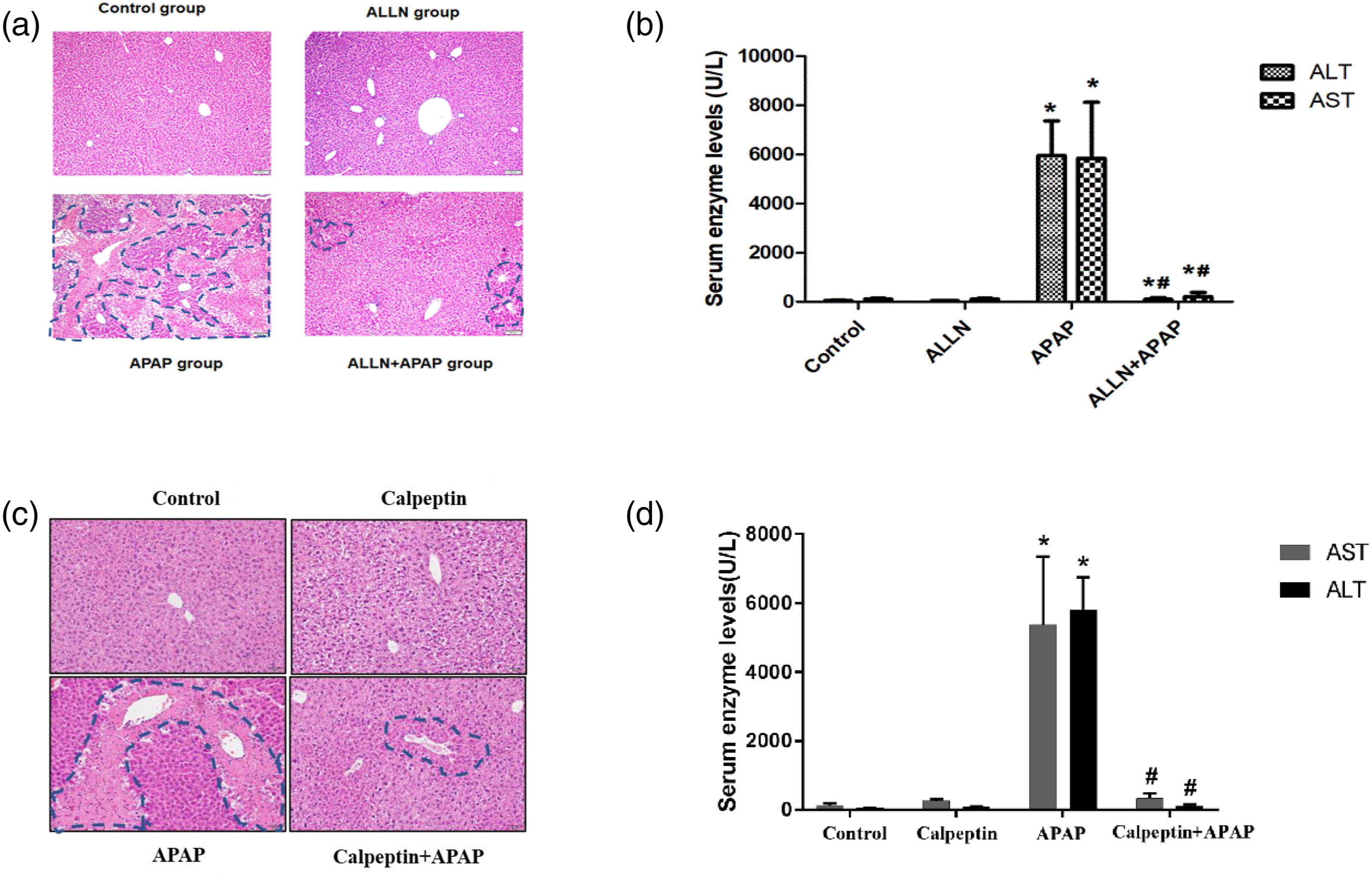
Similarly, calpeptin administration also significantly attenuated APAP-induced liver injury. As shown in Figures 6(c) and 6(d), pretreatment with calpeptin attenuated the increase in ALT and AST activities by nearly 90% and significantly alleviated the hepatocytes necrosis. Taken together, these results support that calpain inhibitors effectively protect against APAP hepatotoxicity.
Discussion
In recent years, mitochondrial dysfunction has been increasingly recognized as a critical event in the initiation and development of APAP hepatotoxicity. Mitochondria are essential organelles for the life and death of eukaryotic cells and participate in aerobic energy production, intermediary metabolism, Ca2+ signaling and apoptosis. 24 Toxic doses of APAP could result in a lowering of cellular ATP levels as well as impaired mitochondrial morphology and function.25,26 More importantly, it is generally recognized that mitochondrial dysfunction is causally linked to the change of mitochondrial dynamics following APAP overdose. 6
In this study, APAP overdose caused severe mice liver injury in a dose-dependent manner, as indicated by the increased serum ALT and AST activities and extensive centrilobular necrosis. Under normal conditions, a dynamic balance between mitochondrial biogenesis and its turnover is strictly regulated. Among them, mitochondrial dynamics is responsible for the recycling of superfluous and damaged mitochondria. 27 Recent findings provide compelling evidence that supports the intrinsic link between mitochondrial dynamics and APAP-induced liver injury. For example, fragmented rod- or sphere-shaped morphological characteristics of mitochondria were readily visualized under APAP treatment. 6 Furthermore, the mitochondrial dynamics related to increased mitochondrial fusion and decreased fission were alleviated by p66Shc knockdown in AML12 cells induced by APAP. 6
To investigate the role and its underlying mechanisms of mitochondrial dynamics in APAP hepatotoxicity, we first examined the effect of APAP on mitochondrial fission/fusion in the liver. The results showed that APAP treatment increased the expression of the full-length of Drp1 and decreased the expression of the full-length of MFn2, suggesting a disruption of mitochondrial fission-fusion machinery and the occurrence of mitochondrial fragmentation. Previous studies demonstrated that changes in mitochondrial dynamics could significantly impact almost all aspects of mitochondrial function, and defects in the large GTPases was involved in the dysfunctional mitochondrial fission/fusion machinery in many human neurological diseases.28,29 For example, glutamate-induced mitochondrial fragmentation observed in a previous study suggested a tipped balance of these processes toward excessive fission that can be due to either enhanced mitochondrial fission, reduced mitochondrial fusion. 30 Our previous research also found that Drp1 in the liver of mice was significantly increased after APAP exposure. 31 Consistent with these reports, TEM analysis in this study found that the mitochondrial morphology and structure of hepatocytes in APAP-treated mice changed significantly, manifested as an increase in the number of mitochondria, the loss of cristae membrane, and swollen and reduced matrix density. Collectively, these results indicated that there existed abnormal mitochondrial dynamics and increased mitochondrial fragmentation in APAP-induced liver injury, which might be mechanistically related to mitochondrial dysfunction.
Mitochondrial fission and fusion machineries are regulated by proteolysis and posttranslational modifications. In this study, an important finding is that APAP exposure induced an obvious cleavage of mitochondrial fusion/fission proteins. Our results demonstrated that 25 kDa fragment along with another 55 kDa fragment of MFn2 and 25 KDa fragment of Drp1 in mice liver at normal condition, which could be significantly enhanced by APAP treatment. Indeed, the decreased full-length of MFn2 is related to its increased cleavage. In the respect, previous study reported that glutamate exposure caused reduction of the full-length of MFn2 through calpain-dependent cleavage of MFn2, 30 which was similar to our results. Furthermore, the inhibition of MFn2 cleavage almost completely alleviates glutamate-induced mitochondrial fragmentation and motor neuronal death. Interestingly, the full-length form and the cleavage fragment of Drp1 were all increased in APAP-treatment mice liver, which supported that Drp1 also underwent cleavage despite the up-regulation of Drp1 expression. Drp1 contains four domains: an N‐terminal GTPase domain, middle domain, variable domain, and C‐terminal GTPase effector domain. 32 The N‐terminal GTPase domain is important in the regulation of Drp1 self‐assembly into dimers and tetramers. 33 It can stand to reason that cleavage of Drp1 somewhere in the middle domain resulting in 25 kDa fragments as shown in our data. Hence, the activation of proteolytic enzymes caused by APAP will inevitably initiate the degradation of Drp1. However, given that the significant up-regulation of Drp1 expression, the degradation of Drp1 in APAP-induced liver injury is not enough to affect its function of promoting mitochondrial fragmentation.
Although an increase in intracellular calcium concentration in hepatocytes is a known consequence of APAP overdose, its importance in the pathogenesis of APAP-induced liver toxicity is not well understood. Sustained elevation of intracellular Ca2+ is deleterious to cells because it can result in activation of hydrolytic enzymes. Among them, Ca2+ -activated neutral proteases, or calpains are believed to mediate the hydrolysis of critical proteins. In the past, calpains were believed to be primarily responsible for the degradation of axonal cytoskeletal proteins such as spectrin, microtubule associated proteins and neurofilaments.34,35 However, increasing researches have revealed that calpains can degrade a variety of cellular proteins and modulate signal transduction pathways that regulate cell shape, adhesion and migration.36–38 Our research proved that APAP administration resulted in a significant increase in the activated form of m-calpain and μ-calpain. In accordance with calpain activation observed in the APAP group, calpastatin of 130 kDa was converted to the lower-molecular-weight breakdown products (mainly 70 kDa) by autolysis, suggesting the activation of calpain degradative pathway in mice liver after the exposure of APAP. Furthermore, this result was further confirmed by calpain activity assay. These results are consistent with a previous report. 39 The roles of calpain in regulating mitochondrial function via hydrolysis critical proteins have been elaborated in many diseases, such as dilated heart failure 40 and diabetic hearts. 41 Considering the above studies and our results, we supposed that calpain activation is at least one of the posttranscriptional mechanisms that contribute to the changes in the levels of large GTPases involved in mitochondrial fission/fusion and dysregulation of mitochondrial dynamics in APAP-induced liver injury.
To identify the exact role of calpain mediating cleavage of mitochondrial fusion/fission proteins in APAP liver injury, we conducted two intervention experiments with ALLN and calpeptin. ALLN pretreatment can significantly attenuate liver injury and inhibit the activation of the calpain degradation system in mice liver after the exposure of APAP. Importantly, this study clearly shown that the dose-dependent cleavage of MFN2 and Drp1 following APAP was significantly inhibited by ALLN and calpeptin. Furthermore, calpain inhibitors effectively prevented the change of mitochondrial dynamics or the occurrence of mitochondrial fragmentation.
It is currently believed that the phosphorylation status of mitochondrial dynamics-related proteins is also involved in the regulation of mitochondrial division. In the respect, fission activity of Drp1 is regulated by its phosphorylation status at multiple amino acid residues, and cytosolic calcium signaling could either increase Drp1 phosphorylation at Ser616 by Ca2+/calmodulin‐dependent kinase II 42 or decreased Drp1 phosphorylation at Ser637 through calcineurin. 43 Interestingly, the activation of calcineurin also requires the activity of calpain. Previous researches reported that mitochondrial fragmentation via dephosphorylation of Drp1 (Ser637) could lead to neuronal death. 23 In the present study, increased dephosphorylation on Ser637 residue and phosphorylation on Ser616 residue of Drp1 was observed in mice liver after APAP treatment. Furthermore, we also observed increased levels of active calcineurin in APAP-treatment mice, while found significantly reduced levels which was inhibited by ALLN or calpeptin.
Several previous studies have also investigated the role of calpain degradation system in APAP liver injury. For instance, another calpain inhibitor, N-CBZ-VAL-PHE-methyl ester (CBZ) was found to significantly increase the survival rate of mice receiving a lethal dose of APAP when it was administered 1 h after APAP. 39 Furthermore, transgenic experiment has revealed that calpastatin overexpression mice exhibited more resistance against APAP hepatotoxicity, as indicated by an attenuated progression of liver injury and a higher survival rate. 44 In this study, our observations are in good agreement with these published papers. Overall, it is very reasonable to speculate that calpain at least partially contribute to APAP-induced mitochondrial dysfunction.
In fact, the activation of hepatocyte proteolytic enzymes during APAP liver injury is not limited to calpain. In a previous study, Cathepsin B activity was significantly increased in liver of C57BL/6 mice after 300 mg/kg APAP treatment. However, inhibition of cathepsin B activity with the inhibitor AC-LVK-CHO could not attenuate APAP-induced liver necrosis. Thus, it is believed that the release of cathepsin B into the cytosol does not contribute to liver injury. 45 Given the different roles of calpain and cathepsin B in regulating the progression of APAP hepatotoxicity, it is reasonable to assume that the role of calpain in APAP hepatotoxicity is specific, which is related to the pathogenesis of APAP -induced mice liver injury.
Of course, most chemical inhibitors generally do not specifically inhibit the activity of a certain proteolytic enzyme. For example, ALLN not only inhibits calpain I and Ⅱ but also proteasome activity, although it is a commonly used as calpain inhibitor. Therefore, in order to exclude the possibility of the proteasome system, we used Calpeptin to conduct a verification experiment. As a calpain-specific inhibitor, Calpeptin binds to the active site of calpain and reversibly inhibits its protease activity. 46 Under normal working concentration, it hardly inhibits the activity of other proteolytic enzymes. As mentioned above, Calpeptin also significantly inhibited the cleavage of mitochondrial dynamics-related proteins, so the results further confirmed the role of calpain in APAP-induced the change of mitochondrial dynamics.
In conclusion, this study has demonstrated that the cleavage of mitochondrial fusion/fission proteins represents an important pathogenic mechanism underlying APAP-induced liver injury. Furthermore, administration of calpain inhibitors could significantly suppress the change of mitochondrial dynamics and alleviate the intoxication of APAP. Thus, our results for the first time provided a mechanistic linkage between calpain activation and mitochondrial dysfunction in APAP-induced acute liver injury. These results suggest that targeting calpain by pharmacological inhibitors could be a new strategy for the prevention and treatment of APAP hepatotoxicity.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Key research and development plan of Shandong Province (2018GSF118013) and National Natural Science Foundation of China (No. 82173552).