
Abstract
This study aimed to investigate the inhibitory effect and mechanism of Cucurbitacin I (Cu I) on apoptosis, oxidative stress, and mitophagy in PC12 cells with glucose and oxygen deprivation/reperfusion (OGD/RP) injury. OGD/RP cell injury model was established by gas anoxic cell incubator and glucose-free medium. The cells were divided into the control group, OGD/RP group, OGD/RP + Cu I group, and OGD/RP + Cu I + 2 µM nuclear factor erythroid 2-related factor 2 (Nrf2) inhibitor ML385 group. The results showed that apoptotic rate and reactive oxygen species (ROS) production were significantly increased in OGD/RP group, which were reversed by Cu I pretreatment. Meanwhile, western blot analysis proved that Cu I inhibited OGD/RP-induced mitophagy, manifested as the decreased expression of PTEN-induced kinase 1 (PINK1) and parkin RBR E3 ubiquitin-protein ligase (Parkin), and light chain 3 (LC3) Ⅱ∕LC3 I, as well as the increased expression of P62. Furthermore, immunofluorescence (IF) staining showed that Cu I reduced the co-localized puncta of LC3 with TOM20 in OGD/RP-induced PC12 cells. Similarly, transmission electron microscope finding is consistent with the IF results. Mechanically, after Cu I and OGD/RP treatments, nuclear Nrf2 expression and the levels of downstream target genes were significantly upregulated compared with OGD/RP alone treatment. Nrf2 inhibition reversed the protective effects of Cu I on OGD/RP-induced injury in PC12 cells. The present study provides evidence of the neuroprotective effect of Cu I unraveling its potential as a potential therapeutic candidate for the treatment of ischemic stroke.
Introduction
Acute ischemic stroke is the most common type of stroke, accounting for about 80% to 90% of all strokes. 1 Over the years, researchers have done a lot of work in the prevention and treatment of acute cerebral infarction, such as positive anticoagulation, thrombolysis, emergency intervention as well as the methods of minimally invasive surgery, and greatly improved the prognosis of patients with acute cerebral infarction. However, a considerable proportion of patients still suffering from varying degrees of sequelae and stroke recurrence. 2 Thrombolytic treatment in the early stage has proved to be one of the effective therapies in acute cerebral infarction. But recanalization happens in about 30% of acute cerebral infarction patients because of thrombolysis therapy or spontaneous to the distal embolus.3,4 Recanalization will be accompanied by the occurrence of ischemia/reperfusion injury. Thus, it is important that study ischemia/reperfusion injury mechanisms and develop new drugs against ischemic damage or find the new role of traditional drugs against ischemic damage. 5 The mechanism of cerebral ischemia/reperfusion injury is mainly associated with the energy exhaustion of neurons, toxicity of excitatory neurotransmitter glutamate, calcium overload, ATP depletion, delayed cell death, free radical damage, the release of different cytokines, depolarization of ischemic penumbra, and inflammatory reaction.6,7 In-depth research according to these pathological process and intervention become the key to avoiding ischemia/reperfusion injury.
Recently, many natural compounds have been identified for their potential anti-ischemic reperfusion injury properties in neurons, such as notoginsenoside R1, 8 eugenol, 9 and curcumin. 10 Cucurbitacins belongs to a group of triterpenoids isolated from plant families such as Cucurbitaceae and Cruciferae, which have been shown to have antipyretic, analgesic, anti-inflammatory, antimicrobial, and antitumor activities.11,12 According to the difference in chemical structure, Cucurbitacins were divided into more than 17 categories, including Cucurbitacin A to T. And Cucurbitacin B, D, E, and I have been extensively reported because of their relative abundance in various plants. Cucurbitacin B (CuB) was reported to exert neuroprotective effects by targeting cofilin and regulating the GR/PLC/PKC and TrkA/Ras/Raf/ERK signaling pathways. 13 Furthermore, cucurbitacin E (CuE) decreased autophagy flux and inhibited parkinsonian toxin 1-methyl-4-phenylpyridinium (MPP(+))-induced cell death in PC12 cells. 14 These results demonstrated that cucurbitacins appeared to have roles in protecting the neuronal injury. Particularly, Cucurbitacin I (Cu I) affects the development of various cancers, including ovarian cancer 15 and colon cancer. 16 However, the antioxidant effects of Cu I in cerebral ischemia injury-induced neurons are currently unknown.
Based on the important action of oxidative stress in the development of oxygen-glucose deprivation/reperfusion (OGD/RP)-which lead to neuronal injury, its strict regulation is a potential master plan for alleviating neuronal injury.17,18 The Nrf2/ARE signaling pathway has emerged as a critical regulator pathway of neuronal oxidative injury and apoptosis.19,20 Target genes of Nrf2 include proteins involved in the regulation of the synthesis of antioxidant proteins, drug-metabolizing enzymes, pentose phosphate pathway enzymes, and enzymes involved in nucleotide syntheses, such as heme oxygenase-1 (HO-1), glutathione-S-transferase (GST), quinone oxidoreductase-1 (NQO1), thioredoxins (Trxs), superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px).21,22 Mitochondria are the site of action of Nrf2-induced cytoprotective gene expression. Nrf2 regulated mitochondrial biogenesis by inhibiting mitochondrial membrane permeability transition pore (mPTP) opening and mitophagy.23,24 Activation of the NrF2/ARE signaling pathway reduces mitochondrial fission and promoted forming of interconnected mitochondrial networks, thereby improving oxidative stress status. Recently, it has been shown that Nrf2 knockout (Nrf2(−/−)) mice that were subjected to ischemia/reperfusion (IR) injury exhibited exacerbated hemispheric and neurological deficit, confirming the role of Nrf2 in overcoming IR-mediated oxidative injury. 25
In this study, we used oxygen-glucose deprivation/reperfusion (OGD/RP) to simulate an ischemia-reperfusion injury of neuronal cells in vitro. And we investigated the mechanism of Cu I of anti-OGD/RP injury from the Nrf2/ARE signaling pathway to further provide a theoretical basis and clinical treatment for stroke.
Materials and methods
Drugs and reagents
Cu I was purchased from Sigma (St. Louis, MO, USA, 238590). ML385, a nuclear factor erythroid 2-related factor 2 (Nrf2) inhibitor, was purchased from MedChem Express (MCE Co. Ltd., Shanghai, China, HY-100523).
Cell culture
The rat pheochromocytoma PC12 cells were purchased from Procell (Wuhan, Hubei, China, #CL-0412). The cells were cultured in high glucose DEME medium (Procell) with 10% fetal bovine serum (Gibco, Paisley, UK, 10099141), 5% horse serum (Gibco, Paisley, UK, 16050122), and 1% penicillin-streptomycin (sv30010, Hyclone, Logan, UT) in a humidified incubator with 5% CO2 and 37°C. The culture medium was replaced at a 2/3 ratio every 2 days. The preliminary experiment was conducted to select the optimal concentration of Cu I, PC12 cells were treated with different concentrations of Cu I (0.1, 0.2, 0.5, 1, 2, 5, and 10 μM) for 24 h and then Cell Counting Kit-8 (CCK-8) assay was performed.
OGD/RP and drug treatment
For our tests, cells were pretreated with 1 μM Cu I for 24 h and exposed to Earle’s balanced salt solution to prepare OGD/RP model. Then, the cells were transferred to an anaerobic chamber containing 5% CO2 and 95% N2 at 37°C for 4 h and then allowed to undergo reoxygenation. After reoxygenation, the cells were placed on a normoxic incubator for 24 h. The cells of the control group were incubated in a normal DMEM medium under the normoxic condition at 37°C in the absence of OGD/RP exposure and Cu I pretreatment. When the Nrf2 inhibitor was introduced, the PC12 cells were preincubated with 2 µM ML385 for 6 h and pretreated with 1 μM Cu I for 24 h before OGD/RP exposure.
Cell viability assay
After different treatments, the cell vitality of PC12 cells was monitored using a Cell Counting Kit-8 (CCK-8, BS350B, Biosharp, Hefei, Anhui, China) according to the manufacturer’s instructions. The absorbance was measured at 450 nm using a spectrophotometer (Bio-Tek, Winooski, VT, USA).
Apoptosis assay
PC12 cell apoptosis was analyzed through Annexin V-FITC following the instructions provided by the manufacturer. Briefly, after washing the cells with PBS), we adapted the cell density to 1.0 × 106 cells/ml. The cells were subsequently suspended in a 150 μl buffering solution. Subsequently, the cells were stained with 10 μg/ml Annexin V-FITC and 5 μl PI at 4°C for 20 min in darkness. Apoptotic cells were evaluated by a BD FACSCelesta™ Flow Cytometer (Becton, Dickinson, and Company, San Jose, CA, USA).
Measurement of reactive oxygen species (ROS) level
The changes in ROS levels in PC12 cells were measured using 2′,7′-dichlorofluorescein-diacetate (DCFH-DA, Sigma, MO, USA, D6883) staining according to the manufacturer’s protocol. The cell precipitation of PC12 cells in each group was harvested and washed with PBS three times. Then the cells were seeded into a 24-well plate (1.0 × 103/well) and stained with 10 μM DCFH-DA for 30 min in the dark. The ROS generation was analyzed by flow cytometry (Bender MedSystems, Vienna, Austria).
Mitochondrial membrane potential measurement
Mitochondrial membrane potential (MMP) of PC12 cells were tested using fluorescent probe JC-1(Nanjing KeyGen Biotech. Co. Ltd. Jiangsu, China, KGA601). PC12 cells were incubated with JC-1 (10 μM) at 37°C for 30 min. Afterward, the cells were rinsed and suspended with 1 × Incubation Buffer. The level of MMP was measured by flow cytometry (Bender MedSystems, Vienna, Austria).
Antioxidant enzymes content measurement
The contents of superoxide dismutase (SOD, ZC-38036) and glutathione peroxidase (GSH-Px, ZC-38196) in the cell culture supernatant of PC12 cells were measured by corresponding Enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s protocol (ZCi Bio, Shanghai, China).
Real-time fluorescence quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated using TRIzol® reagent (Thermo Fisher, Massachusetts, USA, 15596026). SYBR Premix Ex Taq kit (Takara, Bao Biological Engineering, Dalian, China, RR420A) was used following the guidelines. The reverse transcriptional reaction condition was as follows: 95°C for 10 min; 40 cycles of 95°C for 5 s, 60°C for 30 s, and 70°C for 60 s. The sequences of specific miRNA RT primers (Invitrogen, Carlsbad, CA, USA) were as follows: heme oxygenase-1 (HO-1) forward, 5′-TGC TCA ACA TCC AGC T -3′ and reverse, 5′-GCA GAA TCT TGC ACT T-3′, glutamate-cysteine ligase catalytic subunit (GCLC) forward, 5′-CTC CTC ACA GTC ACG GCA TT-3′ and reverse, 5′- TGA ATG GAG ACG GGG TGT TG-3′, modifier subunit (GCLM) forward, 5′-AAG CCA GAC ACT GAC ACA CC-3′ and reverse, 5′- ATC TGG AGG CAT CAC ACA GC-3′, β-actin forward, 5′-CAT GTA CGT TGC TAT CCA GGC-3′ and reverse, 5′-CTC CTT AAT GTC ACG CCA CGA T-3'. The relative gene expression level was determined using the 2-△△Ct method on ABI software, Foster City, CA.
Western blot analysis
Primary antibodies used for Western blot analysis.

Transmission electron microscopy
The formation of autophagosomes in the PC12 cells was observed by Transmission electron microscopy (TEM). Briefly, PC12 cells were fixed with 3% glutaraldehyde for 15 min and post-fixed with 1% osmium tetroxide for 2 h at 4°C. The cells were dehydrated using different concentrations of ethanol (50, 70, 90, 95, and 100% for 20 min, respectively). The cells were then incubated with propanone for 2 h and embedded with Ep812 resin. The blocks were sliced with a Leica EM UC7 and sample sections were stained with uranium acetate-lead citrate. A JEM-1400Flash transmission electron microscopy (JEOL, Tokyo, Japan) was used to examine.
Immunofluorescence
The co-expression of the translocase of outer membrane 20 kDa subunit (TOM20) and light chain 3 (LC3) in PC12 cells was estimated by Immunofluorescence (IF). Briefly, PC12 cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.01% Triton X-100 for 10 min, and then blocked with 5% normal goat serum for 1 h at room temperature. The cells were incubated with an anti-TOM20 and anti-LC3 antibody overnight at 4°C, washed with PBS, and then incubated with a FITC-conjugated secondary antibody in the dark for 2 h at room temperature. The nuclei of the cells were stained with DAPI. All samples were analyzed using fluorescence microscopy (Nikon TE2000 microscope, Nikon, Tokyo, Japan). Fluorescence intensity was performed with ImageJ software.
Statistics analysis
The data were represented as means ± standard deviation (SD). Statistical analysis was performed with the SPSS software (version 19.0, SPSS Inc., Chicago, IL, USA). One-way analysis of variance (ANOVA) followed by Tukey’s-b post hoc test was used for comparison between groups. p-value < 0.05 was considered statistically significant.
Results
Cu I rescued PC12 cells from OGD/RP-induced PC12 cell apoptosis
We first determined the optimal concentration of Cu I. Cell viability of PC12 cells was assessed after treatments with different concentrations of Cu I. As shown in Figure 1(a) and 1(b), 1 μM Cu I was optimal for the promotion of cell viability which was selected for further study (1 μM vs Con, 1 μM vs OGD/RP, p < 0.01). To investigate the effects of Cu I on OGD/RP-induced cell viability in PC12 cells, 1 μM Cu I was used to pretreat PC12 cells for 24 h before OGD/RP exposure. CCK-8 assay showed that the viability of OGD/RP-induced PC12 cells was decreased compared with that in control cells, which was significantly rescued by Cu I pretreatment (OGD/RP vs Con, 1 μM vs OGD/RP, p < 0.001; Figure 1(b)). Additionally, in OGD/RP-induced cells, cell apoptosis was increased compared with that of control cells (OGD/RP vs Con, p < 0.001; Figure 1(c)). However, pretreatment with Cu I for 24 h significantly decreased the cell apoptosis compared with that in OGD/RP-induced cells from the flow cytometry detection assay (Cu I vs OGD/RP, p < 0.05; Figure 1(c)) and Western blot analysis of apoptosis regulators, such as Bax (Cu I vs OGD/RP, p < 0.05; Figures 1(d)-(e)), Bcl-2 (Cu I vs OGD/RP, p < 0.01; Figures 1(d)-(e)), and cleaved-caspase3 (Cu I vs OGD/RP, p < 0.01; Figures 1(d)-(e)). These results suggest that OGD/RP significantly induced PC12 cell apoptosis, and Cu I could protect PC12 cells from OGD/RP insult. Cu I rescued PC12 cells from OGD/RP-induced cell apoptosis. (a and b) Cu I enhanced OGD/RP-impaired cell viability. Cell viability was measured using the CCK-8 assay in cells pretreated with indicated Cu I concentrations for 24 h followed by OGD/RP treatment. (c) Cu I suppressed the apoptosis of OGD/RP-induced PC12 cells. Apoptosis induced by OGD/RP was determined with Annexin V-FITC/PI staining followed by Flow cytometry analysis in PC12 cells. Percentages of apoptotic cells are shown. (d and e) Cu I pretreatment attenuated the accumulation of apoptosis-associated proteins in OGD/RP-induced PC12 cells. After 1 μM Cu I pretreatment and OGD/RP incubation for the indicated time, the levels of Bax, Bcl-2, caspase3, and cleaved-caspase3 protein expression levels were analyzed by Western blot. β-actin was used as the loading control. Data are expressed as fold changes ±SD. *p < 0.05 (vs Control), **p < 0.01 (vs Control), ***p < 0.001 (vs Control), #p < 0.05 (vs OGD/RP), ##p < 0.01 (vs OGD/RP).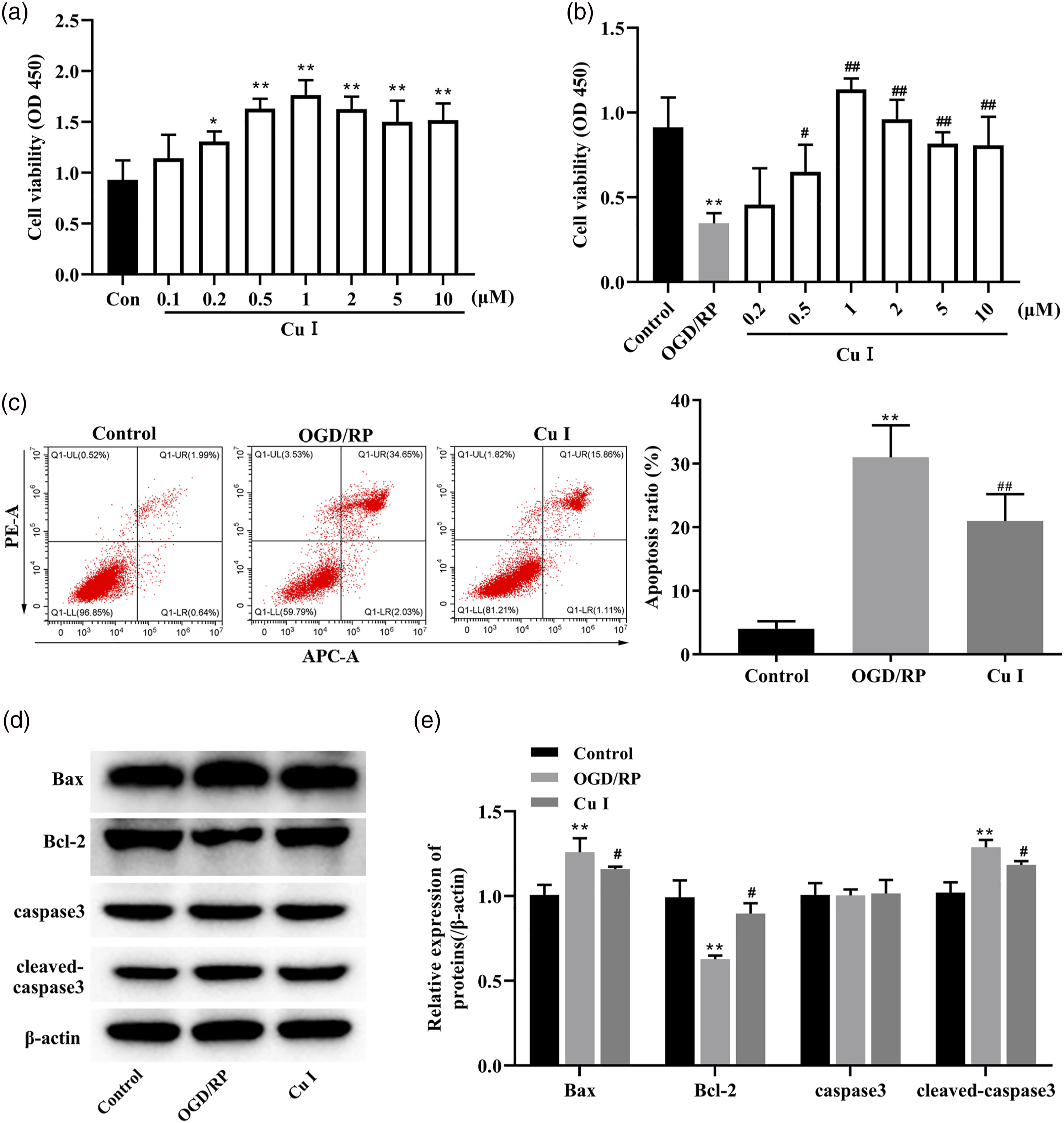
Cu I prevented the accumulation of oxidative stress in OGD/RP-induced PC12 cells
To determine whether Cu I can inhibit the oxidative stress in OGD/RP-induced PC12 cells, cells were pretreated with 1 μM Cu I for 24 h followed by OGD/RP treatment. From the flow cytometry of DCFH-DA as a ROS sensor dye, OGD/RP treatment increased intracellular ROS levels compared with that in control cells (OGD/RP vs Con, p < 0.001; Figure 2(a)). Otherwise, pretreatment with Cu I dramatically diminished the ROS levels compared with those in OGD/RP-induced PC12 cells (Cu I vs OGD/RP, p < 0.001; Figure 2(a)). Of note, oxidative stress usually manifests as the collapse of the mitochondrial membrane potential (MMP). Out data proved that OGD/RP exposure resulted in the dissipation of the MMP, which was stabilized by Cu I pretreatment (OGD/RP vs Con, p < 0.001; Cu I vs OGD/RP, p < 0.01; Figure 2(b)). Since antioxidant proteins, including SOD and GSH-Px, have preventive functions against oxidative stress, we further examined the expression levels of these proteins in each group of cells. An obvious depletion in the content of SOD (OGD/RP vs Con, p < 0.01; Figure 2(c)) and GSH-Px (OGD/RP vs Con, p < 0.01; 2D) were found after OGD/RP exposure. As expected, pretreatment with Cu I markedly increased the expression of SOD (Cu I vs OGD/RP, p < 0.05; Figure 2(c)) and GSH-Px (Cu I vs OGD/RP, p < 0.05; Figure 2(c)) compared with those in OGD/RP-induced cells (Figures 2(c) and (d)). The data above suggested that Cu I reduced oxidative stress in PC12 cells induced by OGD/RP treatment. Cu I prevented the accumulation of oxidative stress in OGD/RP-induced PC12 cells. PC12 cells were pretreated with 1 μM Cu I for 24 h followed by OGD/RP treatment. (a) Cu I pretreatment inhibited reactive oxygen species (ROS) production in OGD/RP-induced PC12 cells. Intracellular ROS production was measured by DCFH-DA staining. (b) Cu I enhanced mitochondrial membrane potential (MMP) of OGD/RP-induced PC12 cells. MMP was tested by JC-1 probe. (c and d) Cu I promoted antioxidant enzymes expression in OGD/RP-induced PC12 cells. The contents of superoxide dismutase (SOD) and glutathione-peroxidase (GSH-Px) in cell culture supernatant were measured by corresponding ELISA kits. Data are expressed as fold changes ±SD. **p < 0.01 (vs Control), ***p < 0.001 (vs Control), #p < 0.05 (vs OGD/RP), ##p < 0.01 (vs OGD/RP), ###p < 0.001 (vs OGD/RP).
Cu I inhibited PTEN-induced kinase 1 (PINK1)/parkin RBR E3 ubiquitin-protein ligase (Parkin) dependent mitophagy in OGD/RP-induced PC12 cells
We next examined the effects of Cu I on mitophagy in OGD/RP-induced PC12 cells. Western blot analysis was performed to check the expressions of autophagy-related proteins. We found a significant increase in LC3 Ⅱ∕LC3 I, PINK1, and Parkin in OGD/RP-induced cells, whereas the expression of P62 was significantly down-regulated compared with the control group (OGD/RP vs Con, p < 0.001; Figures 3(a)-(f)). And Cu I pretreatment reversed these proteins' expression (LC3 Ⅱ∕LC3 I, Cu I vs OGD/RP, p < 0.01; ⅠPINK1, Parkin, and P62, Cu I vs OGD/RP, p < 0.001; Figures 3(a)-(f)). Meanwhile, IF with anti-LC3 and anti-TOM20 antibodies was used to test the co-localization of LC3 puncta with mitochondria. As shown in Figure 3(g), OGD/RP treatment enhanced co-localization of TOM20 and LC3, which was significantly blocked by Cu I (OGD/RP vs Con, p < 0.01; Cu I vs OGD/RP, p < 0.05). In addition, autophagosomes (shown as yellow arrows) in PC12 cells were further detected by TEM and similar results were obtained (Figure 3(h)). The above results demonstrated that Cu I pretreatment protected PC12 cells from autophagy caused by OGD/RP treatment. Cu I inhibited PINK1/Parkin-dependent mitophagy in OGD/RP-induced PC12 cells. PC12 cells were pretreated with Cu I (1 μM) for 24 h followed by OGD/RP treatment. (a–f) Cu I pretreatment regulated the expression of autophagy-associated proteins in OGD/RP-induced PC12 cells. Western blot analysis of LC3, P62, ⅠPINK1, and Parkin expression. β-actin was used as the loading control. (g) Cu I hindered mitophagy of OGD/RP-induced PC12 cells. Representative images of immunofluorescence (IF) of LC3 (red) and TOM20 (mitochondria marker, green) in PC12 cells. Scale bar = 100 μm. LC3 + TOM20-stained area/cell area was measured. (h) Transmission electron microscopy (TEM) was used to assess mitophagy. Yellow arrowhead indicates autophagy of mitochondria. Scale bar = 500 nm. **p < 0.01 (vs Control), ***p < 0.001 (vs Control), #p < 0.05 (vs OGD/RP), ##p < 0.01 (vs OGD/RP), ###p < 0.001 (vs OGD/RP).
Cu I promoted Nrf2/ARE signaling pathway in OGD/RP-induced PC12 cells
Next, we investigated whether Nrf2/ARE signaling is involved in the anti-OGD/RP effect of Cu I in PC12 cells. PC12 cells were pretreated with Cu I after OGD/RP, and the change in Nrf2 was evaluated in the nucleus and cytoplasm. As shown in Figures 4(a)-(c), the expression of Nrf2 in the nucleus was significantly decreased in the OGD/RP group (OGD/RP vs Con, p < 0.001). As expected, Cu I pretreatment promoted OGD/RP-reduced nuclear translocation of Nrf2 (Cu I vs OGD/RP, p < 0.01; Figures 4(a)-(c)). In addition, HO-1, GCLC, and GCLM are known to be the targeted genes of Nrf2. As shown in Figure 4(d), PCR-qPCR analysis showed that mRNA levels of HO-1 (OGD/RP vs Con, p < 0.05), GCLC (OGD/RP vs Con, p < 0.01), and GCLM (OGD/RP vs Con, p < 0.01) are significantly decreased after OGD/RP, which were slightly alleviated in transcription caused by Cu I pretreatment (Cu I vs OGD/RP, p < 0.05). These findings further indicated that the anti-OGD/RP effect of Cu I was dependent on the activation of the Nrf2/ARE pathway. Cu I promoted Nrf2/ARE signaling pathway in OGD/RP-induced PC12 cells. PC12 cells were pretreated with 1 μM Cu I for 24 h followed by OGD/RP treatment. (a–c) Cu I stimulated Nrf2 nuclear translocation of OGD/RP-induced PC12 cells. Western blot analysis of cytoplasmic Nrf2 and nuclear Nrf2 expression. Lamin B and β-actin were used as markers for nucleus and cytoplasm, respectively. (d) Cu I promoted the expression of Nrf2/ARE signaling downstream target genes in OGD/RP-induced PC12 cells. The mRNA levels of heme oxygenase-1 (HO-1), glutamate-cysteine ligase catalytic subunit (GCLC), and modifier subunit (GCLM) were tested by RT-qPCR analysis. Data are expressed as fold changes ±SD. *p < 0.05 (vs Control), **p < 0.01 (vs Control), #p < 0.05 (vs OGD/RP), ##p < 0.01 (vs OGD/RP).
The inhibition of the Nrf2/ARE pathway reversed the inhibitory effect of Cu I on OGD/RP induced oxidative stress in PC12 cells
To further verify the proposed functional correlation of signaling cascade, we assessed whether the Nrf2 inhibitor ML385 could influence the therapeutic efficacy of Cu I on OGD/RP injury. As shown in Figures 5(a)-(d), Cu I promoted Nrf2 nuclear accumulation (Cu I vs OGD/RP, p < 0.001), and the RNA levels of its downstream target genes HO-1 (Cu I vs OGD/RP, p < 0.001), GCLC (Cu I vs OGD/RP, p < 0.05), and GCLM (Cu I vs OGD/RP, p < 0.01). As expected, Nrf2 inhibitor ML385 significantly blocked these changes, including the decreased expression of nuclear Nrf2 (Cu I + ML385 vs Cu I, p < 0.01), HO-1 (Cu I + ML385 vs Cu I, p < 0.05), GCLC (Cu I + ML385 vs Cu I, p < 0.05), and GCLM (Cu I + ML385 vs Cu I, p < 0.05) in PC12 cells (Figures 5(a)-(d)). Furthermore, after the pretreatment of ML385 and Cu I followed by OGD/RP, the ROS level in PC12 cells was detected, and the results showed Nrf2 inhibitor ML385 increased ROS level compared with this in the Cu I-treated cells followed by OGD/RP (Cu I + ML385 vs Cu I, p < 0.05; Figure 6(a)). And the MMP level showed the opposite results (Cu I + ML385 vs Cu I, p < 0.05; Figure 6(b)). Meanwhile, our results manifested that the contents of antioxidant enzymes SOD (Cu I + ML385 vs Cu I, p < 0.05; Figure 6(c)) and GSH-Px (Cu I + ML385 vs Cu I, p < 0.05; Figure 6(d)) were reduced by Nrf2 ML385 in Cu I-treated PC12 cells followed by OGD/RP. These data indicated the implication of Nrf2/ARE signaling in the protective role of Cu I against OGD/RP-induced oxidative stress. Nrf2 inhibitor blocked the increased activity of the Nrf2/ARE signaling pathway which was induced by Cu I in PC12 cells. PC12 cells were pre-incubated with 2 µM ML385 (Nrf2 inhibitor) for 6 h and pretreated with 1 μM Cu I for 24 h before OGD/RP exposure. ML385 suppressed nuclear Nrf2 and its downstream genes induced by Cu I in PC12 cells. (a–c) Western blot analysis of cytoplasmic Nrf2 and nuclear Nrf2 expression. Lamin B and β-actin were used as markers for nucleus and cytoplasm, respectively. (d) The mRNA levels of HO-1, GCLC, and GCLM were tested by RT-qPCR analysis. Data are expressed as fold changes ±SD. *p < 0.05 (vs Control), **p < 0.01 (vs Control), #p < 0.05 (vs OGD/RP), ##p < 0.01 (vs OGD/RP). The inhibition of the Nrf2/ARE pathway reversed the inhibitory effect of Cu I on OGD/RP-

Nrf2/ARE pathway mediated the inhibitory effect of Cu I on OGD/RP induced mitophagy in PC12 cells
Finally, we examined whether Nrf2 inhibition affects autophagy in Cu I-treated PC12 cells. Western blot analysis proved that the Cu I-inhibited LC3 Ⅱ/LC3 I (Cu I vs OGD/RP, p < 0.001; Cu I + ML385 vs Cu I, p < 0.05), PINK1 (Cu I vs OGD/RP, p < 0.001; Cu I + ML385 vs Cu I, p < 0.05), and Parkin (Cu I vs OGD/RP, p < 0.001; Cu I + ML385 vs Cu I, p < 0.01) expression were significantly reversed by ML385 treatment in PC12 cells (Figures 7(a)-(f)). Meanwhile, the expression levels of p62 (Cu I vs OGD/RP, p < 0.001; Cu I + ML385 vs Cu I, p < 0.01) showed the opposite trend of changes (Figures 7(a)-(f)). Moreover, the decreased co-expression of autophagy protein LC3 and mitochondrial protein TOM20 in Cu I-treated PC12 cells was reversed by ML385 treatment (Cu I + ML385 vs Cu I, p < 0.05; Figure 7(g)).TME representative images were in line with the results from IF (Figure 7(h), mitophagy was shown as yellow arrows). These findings further indicated that Nrf2 inhibition blocked the protective effect of Cu I on OGD/RP-induced PC12 cells. Nrf2/ARE pathway mediated the inhibitory effect of Cu I on OGD/RP induced mitophagy in PC12 cells. PC12 cells were pre-incubated with 2 µM ML385 (Nrf2 inhibitor) for 6 h and pretreated with 1 μM Cu I for 24 h before OGD/RP exposure. (a–f) The ML385-induced autophagy-related proteins expression augmentation in Cu I-treated PC12 cells. Western blot analysis of LC3, P62, ⅠPINK1, and Parkin expression. β-actin was used as the loading control. (g) Nrf2 inhibition exacerbated mitophagy in Cu I-treated cells. Double immunofluorescence (IF) images of LC3 (red) and TOM20 (green) in PC12 cells. Scale bar = 100 μm. LC3 + TOM20-stained area/cell area was measured. (h) Transmission electron microscopy (TEM) was used to test mitophagy. Yellow arrowhead indicates autophagy of mitochondria. Scale bar = 500 nm. Data are expressed as fold changes ±SD. **p < 0.01 (vs Control), ***p < 0.001 (vs Control), #p < 0.05 (vs OGD/RP), ###p < 0.001 (vs OGD/RP), &p < 0.05 (vs OGD/RP + Cu I), &&p < 0.01 (vs OGD/RP + Cu I).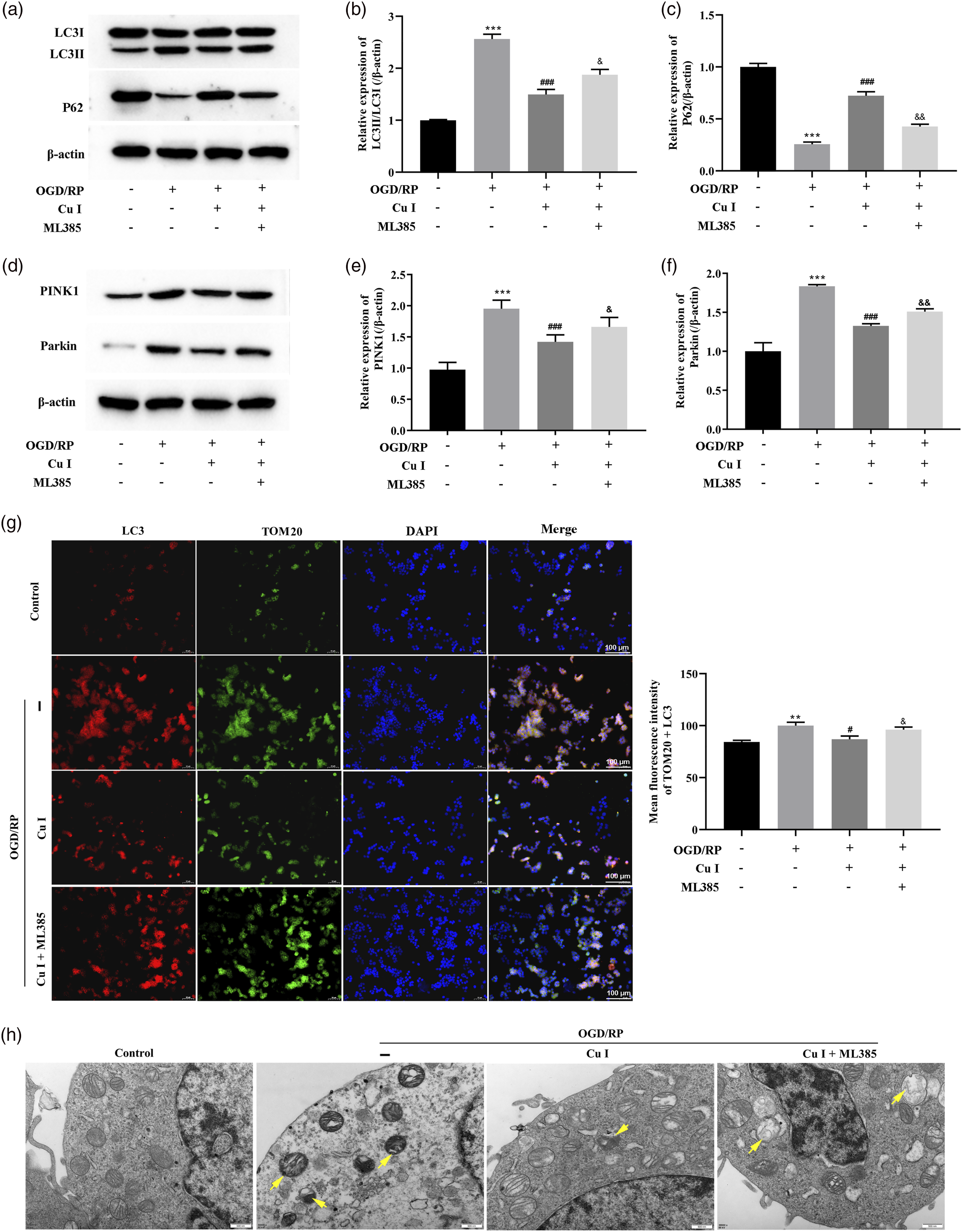
Discussion
Cu I has been widely investigated in anti-cancer,
26
management of cardiomyocyte hypertrophy,
27
and treatment of metabolic diseases.
28
In addition to these effects, we demonstrated that Cu I had remarkable effects on anti-OGD/RP injury in neurons, including resistance to apoptosis, oxidative stress, and autophagy accumulation. Figure 8 The mechanistic scheme of Cu I in OGD/RP-induced injury. Cu I protected against oxidative stress, autophagy, and apoptosis in OGD/RP-induced PC12 cells by activating the Nrf2/ARE pathway.
When the cerebral infarcted, the regional center of cerebral blood vessels dominant will form the core area and the surrounding ischemic penumbra, in which neuronal necrosis is are mainly in the former, while the latter is composed of neuronal apoptosis and is the key point to protect. 29 The expression of apoptosis-related proteins, such as Bcl-2, Bax, and cleaved-caspase3 was changed after cerebral ischemia. 30 Moreover, oxidative stress has been proved to induce apoptosis of nerve cells. ROS produced by oxidative stress can mediate apoptosis. 31 The relevant study showed that ROS production significantly increased after cerebral ischemia/reperfusion injury. 32 Following these, our study also showed that OGD/RP treatment inhibited cell viability, as well as enhanced apoptosis and oxidative stress of PC12 cells. Interestingly, Cu I pretreatment during OGD/RP period significantly antagonized OGD/R-triggered cell injury.
Autophagy, which resides in almost all cells that have mitochondria, is a traditional conserved homeostasis process. 33 Virtually, autophagy occurs at low basal levels in cells to execute homeostatic functions, such as organelles and protein turnover. It is up-regulated rapidly when intracellular nutrition and energy are deficient, like hypoxia, starvation, and the depletion of growth factors. Autophagy also can induce programmed cell death and is termed type II programmed cell death when it is inappropriately activated. Besides that, autophagy can lead to apoptosis because of their crosstalk. At present, the exact role of autophagy in ischemic brain injury remains ambiguous. The evidence suggested that OGD/RP exposure successfully activated autophagy. And the elevated autophagy might be a scathing factor in OGD/RP injury of neurons. 34 In contrast, another study has demonstrated that enhanced autophagy had neuroprotective effects in focal cerebral ischemia in rats. 35 In our study, Cu I inhibited OGD/RP-induced autophagy, indicated by decreases in LC3 Ⅱ/LC3 I protein level and accumulation of p62 in PC12 cells. Notably, our findings are different from the previous study which suggested Cucurbitacin enhanced the accumulation of autophagosomes and cell death in a caspase-independent manner. 36
PINK1 is present in the normal outer mitochondrial membrane and could be degraded by protein hydrolases. 37 When a mitochondrial injury occurs, proteolytic enzyme activity was inhibited, resulting in the massive accumulation of PINK1 on the mitochondrial membrane. 37 PINK1 induced the translocation of Parkin from the cytoplasm to the mitochondria, which was a key step in the canonical mitophagy pathway.37,38 Therefore, changes in PINK1/Parkin levels reflected the degree of mitophagy in the intracellular. A previous study indicated that PINK1/Parkin-dependent mitophagy played a role in OGD/R-induced neuronal injury. 39 SIRT1-mediated PINK1/Parkin axis was activated by Apelin-36 treatment with the downregulation of p62 and upregulation of LC3 II and Beclin1 in OGD/R-induced HT22 cells. 39 We proved that Cu I antagonized OGD/RP-triggered neuronal injury, which might be mediated, at least in part, via inhibition of PINK1/Parkin-dependent mitophagy.
Nrf2/ARE signaling pathway is involved in the maintenance of the redox balance under a stress state, and is a potential target for the treatment of oxidative stress-related diseases. Nrf2 is a transcription factor that combines with ARE and induces the expression of various genes. 40 In the nervous system, Nrf2 mainly regulates downstream HO-1, NADPH quinone oxidoreductase-1 (Nqo1), and glutathione-S-transferases (GSTs), thereby protecting nerve cells from oxidative activity. 41 Several studies have confirmed that highlight the protective role of the Nrf2/ARE pathway in cerebral ischemia injury, as it inhibits oxidative stress and neuroinflammation.25,42 Panax notoginseng saponins (PNS), 43 morroniside (MOR), 44 and ginsenoside Rg1 45 protected nerve cells against OGD/RP injury via regulating the Nrf2/ARE/HO-1 pathway. In a cerebral ischemia-reperfusion rat model, levocarnitine relieved cerebral nerve injury and oxidative stress by activating the Nrf2/ARE signaling pathway. 46 Our findings also confirmed that inhibition of the Nrf2/ARE signaling pathway reversed the neuroprotective effects of Cu I pretreatment on OGD/RP-induced PC12 cells.
This study had limitations. First, this research was performed on neuron-like rat pheochromocytoma cell line PC12. The effects we observed could be cell line-specific, although it is notable that Cu I inhibited OGD/RP-induced oxidative stress, mitophagy, and apoptosis in PC12 cells. Second, it would be interesting to extend the study by employing in vivo models to evaluate the neuroprotective effects of Cu I on OGD/RP injury.
Conclusions
In conclusion, our findings showed Cu I protected against oxidative stress, autophagy, and apoptosis in OGD/RP-induced PC12 cells. The mechanism of neuroprotective effects of Cu I was possibly through activating the Nrf2/ARE pathway. The present study provides strong evidence in support of Cu I as a promising therapeutic strategy for the treatment of ischemic stroke.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Research Fund for youth projects (grant number 2020-QYY-6, China).