
Abstract
Anesthesia may induce neuronal tau phosphorylation and neurotoxicity in the developing brain. Apolipoprotein E (ApoE) may play a protective role in neuronal activity and injury repair, whereas its 18-kDa fragments are reported to induce neurodegeneration and neuroinflammation in Alzheimer’s disease patients. We aimed to test the hypothesis that differences in 18-kDa ApoE fragment levels, but not full-length ApoE, in primary neurons contribute to differences in tau phosphorylation and neuroinflammation with or without sevoflurane administration. Neurons extracted from wild-type (WT), ApoE knockout (ApoE-KO), and ApoE ε3-and ε2-targeted replacement (ApoE ε3, ApoE ε2) mice were divided into control and sevoflurane groups. Neurons in the sevoflurane group were treated with 21% O2, 5% CO2, and 4.1% sevoflurane, whereas those in the control group were treated with 21% O2 and 5% CO2 only on day 5 of neuronal culture. ApoE mRNA, full-length ApoE, 18-kDa ApoE fragments, Tau-PS202/PT205 (AT8), Tau-PSer396/404 (PHF1), tumor necrosis factor (TNF)-α, and interleukin (IL)-6 and IL-1β levels were measured. The data showed that sevoflurane-induced AT8 and PHF1 increases, and TNF-α, IL-6, and IL-1β increases in WT or ApoE ε3 neurons (both expressing full-length and 18-kDa fragmented ApoE) could be mitigated in ApoE ε2 (only expressing full-length ApoE), but not in ApoE-KO neurons, indicating that differences in 18-kDa ApoE fragments, but not full-length ApoE, in primary mouse neurons contributed to differences in tau phosphorylation and neuroinflammation with or without 4.1% sevoflurane administration.
Keywords
Background
Sevoflurane is a common inhalational anesthetic used in pediatric anesthesia. 1 However, long-term or repeated exposure to sevoflurane before the age of 3–4 years has been shown to increase the likelihood of future learning and memory deficits,2-4 although conflicting evidence exists.5,6 Furthermore, several preclinical studies have shown that inhalation anesthetics might cause cognitive impairment, brain structural abnormalities, neuroinflammation, apoptosis, synaptic insufficiency, and other adverse effects in neonatal animals and primary cultured cells.1,7-9 A previous study found that 4 h of treatment with 21% O2, 5% CO2, and 4.1% sevoflurane on day 5 of neuronal culture might cause neuroinflammation and tau protein hyperphosphorylation in vitro. 10 The mechanism of the upstream process, on the other hand, is not well understood.
Apolipoprotein E (ApoE) is a key component of lipoproteins that is mainly produced and secreted by astrocytes, but under certain conditions, it can also be expressed in neurons.11-13 According to previous studies, ApoE might play roles in protection and injury repair in the central nervous system (CNS) and help in the removal of beta-amyloid (Aβ) from the brain.14,15 ApoE has three different isoforms in humans: ε2, ε3, and ε4. 16 The ApoE ε3 allele, which encodes the ε3 isoform, is defined as the “wild-type” (WT) allele and is the most common in the population, with an allele frequency of 0.7–0.8. ApoE ε2 and ApoE ε4 are less common, with allele frequencies of 0.05–0.1 and 0.1–0.15, respectively.13,17 The three alleles differ by only two nucleotides, resulting in a protein alteration of two amino acids at positions 112 and 158. ApoE ε2 contains Cys112 and Cys158, ApoE ε3 contains Cys112 and Arg158, and ApoE ε4 contains Arg112 and Arg158. 11 Amino acid changes within the subtypes lead to differences in protein stability; the ApoE ε4 isoform is the most unstable, followed by ApoE ε3, while ApoE ε2 is the most stable.14,18 Moreover, ApoE ε3 and ApoE ε4 are degraded by proteases to produce short fragments in the human brain. 19 It has been reported that tau hyperphosphorylation, Aβ plaque formation, and neurofibrillary tangle (NFT) formation are directly correlated with specific neurotoxic ApoE fragments, for example, an 18-kDa N-terminal fragment, which could give rise to neurocognitive impairment and the initiation of neurodegeneration in patients with Alzheimer’s disease (AD).11,14,19
In the present study, we first examined the association between the increase in total ApoE and the expression levels of inflammatory factors and phosphorylated tau protein after sevoflurane was administered to primary hippocampal neurons extracted from WT or ApoE knockout (ApoE-KO) mice. To further examine whether full-length ApoE or 18-kDa ApoE fragments play a role in the presence and severity of neurotoxicity in neurons treated with sevoflurane, we employed ApoE ε3-targeted replacement (ApoE ε3) and ApoE ε2-targeted replacement (ApoE ε2) mice in the following study. Taken together, the findings of these studies support the hypothesis that differences in 18-kDa ApoE fragments, but not full-length ApoE, in primary mouse neurons contribute to differences in tau phosphorylation and neuroinflammation with or without 4.1% sevoflurane administration.
Materials and methods
Primary neurons culture and treatment
Adult WT and ApoE-KO C57BL/6J mice were obtained from Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China), and ApoE ε3 and ApoE ε2 C57BL/6J mice were purchased from Shanghai Model Organisms Center (Shanghai, China). The Institutional Animal Care and Use Committee of Tianjin Medical University General Hospital (China; clearance number 2018-X6-11) preapproved all animal experimental protocols. The number of animals used in the research was minimized as far as possible.
Anesthetized the mice by inhalation of 3% sevoflurane and 60% oxygen for 1–2 min, the animals were euthanized by decapitation at 15 days’ gestation. During the subsequent protocol, we ensured that the environment was as sterile as possible. Using sterile dissecting scissors and forceps, we created an opening on the mid-ventral side of the pregnant mouse to completely reveal the body cavity. The uterus was opened, and the mouse pups were carefully removed using sterile forceps. We then decapitated the pups with fresh sterile scissors and placed the removed head on sterile gauze under a dissecting microscope. Using eye scissors, the craniums of the pups were opened from the back of the neck to the nose by inserting one tip of the scissors into the vertebral foramen and then proceeding anteriorly. Sterile tweezers were used to carefully remove the entire brain, which was separated into two hemispheres using a scalpel. The hippocampus is a curved structure under the cortex covered by a layer of white meninges. After dissection, we gently lifted each hippocampus with sterile tissue forceps and transferred it to a small tissue culture dish with precooled DMEM. The hippocampus was repeatedly and gently minced with eye scissors and transferred to a glass garden containing 2 mL DMEM. The petri dish was supplemented with 2 mL 0.25% trypsin, which was digested in an incubator at 37°C for 30 min. The tissue debris and digestive fluid were transferred to a centrifuge tube; the same volume of digestive terminating fluid (DMEM + 10% FBS; Gibco, USA) was added and the tube was gently shaken 15 times. Every time the tissue completely settled at the bottom of the tube; it was shaken again. The suspension was then filtered through a 60-μm sieve. After 1 min, the supernatant of the single-cell suspension was collected and mixed with the appropriate digestive termination solution. A volume of 1.5 mL was inoculated into each pore of a six-well Petri plate. A microscope was used to count the cells and ensure a density of 7×105 cells per well. Neuron growth was observed after 4 h of culture by replenishing the neuronal culture medium (Neurobasal-A + 2% B27; Engreen, Beijing, China). Neuronal growth media were replaced every 3 days, and half of the old media was replaced with the same volume of fresh neurobasal culture media each time.
On the 5th day, as described by Lu et al., the neurons of the sevoflurane group were laid on a closed resin box (20 cm × 15 cm × 7 cm) and treated with 21% O2, 5% CO2, and 4.1% sevoflurane (2 minimum alveolar concentration [MAC]) for 4 h in a 37°C incubator. 20 The control group only received 21% O2 and 5% CO2 for 4 h in the same environment, while the concentrations of O2, CO2, and sevoflurane were monitored constantly using a gas monitor (Vamos; Drager Medical AG & Co. KgaA, Germany)
Primary neuron harvesting
Following sevoflurane or control treatment, neurons from each group were immediately extracted for biochemical analyses. The culture medium was removed from the Petri dishes and cleaned three times with phosphate-buffered saline (PBS). After addition of 1 mL pre-cooled protein extract, neurons were gently scraped from the bottom of the culture jar using a cell curette. Finally, the lysates were collected and centrifuged at 12,000 rpm for 10 min.
Cell Counting Kit-8 (CCK-8) cell viability assay
CCK8 was used in accordance with the manufacturer’s instructions. When the sevoflurane or control treatment was finished, the original culture medium in each well was replaced with 90 μL serum free medium, and 10 μL CCK8 reagent (cat. no. CK04; Dojindo Molecular Technologies, Inc.) was immediately added, followed by a 2 h incubation at 37°C in the dark. To test cell viability, the absorbance at 450 nm was measured using a VERSAMax microplate reader (Molecular Devices, LLC). The control group’s viability was assumed to be 100%, and the results of the other groups were computed as a percentage of the control group.
Protein quantification
The total protein content was determined using a bicinchoninic acid protein assay kit (Pierce, Iselin, NJ, USA).
Real-time polymerase chain reaction
A spectrophotometer was used to measure the amount of total extracted RNA. An all-in-one first-strand cDNA synthesis kit (Cat. No. AORT-0050; Gene Copoeia, USA) was used to convert 500 ng mRNA from each group into cDNA as a template. To amplify the cDNA product, the iQTM5 system and Real Master Mix (SYBR Green, ThermoFisher Scientific, USA) were used. To quantify the relative mRNA levels, the 2−ΔΔCq technique was used. The primer sequences were as follows: ApoE, reverse primer 5ʹ-CATGTCTTCCACTATTGGCTCG-3ʹ and forward primer 5ʹ-GACCCAGCAAATACGCCTG-3ʹ; GAPDH, reverse primer 5ʹ-AGGTCGGTGTGAACGGATTTG-3ʹ and forward primer 5ʹ-TGTAGACCATGTAGTTGAGGTCA-3ʹ.
Western blot analysis
Full-length ApoE, 18-kDa ApoE fragments, AT8, and PHF1 expression in neurons were detected by western blotting. SDS-polyacrylamide gels (standard XT 4%–12% double TIS gel (Bio-Rad, USA) or 4%–20% double TIS polyacrylamide gel (Cat. No. M00655, Gen Script Biotech Corp., Nanjing, China) were loaded and subjected to electrophoresis. The gel was transferred to a polyvinylidene fluoride membrane (Millipore, USA), which was then blocked in 5% skimmed milk powder before being probed with specific primary antibodies against full-length ApoE and 18-kDa ApoE fragment (Cat. No. 178479, 1:4,000; Calbiochem, Germany), AT8 (Cat. No. MN1020, 1:2,000; Thermo Fisher Scientific, USA), PHF1 (Cat. No. 3Ab184951, 1:1,000, Abcam, UK) and β-actin (Cat. No. A5060, 1:2,000; Sigma, USA). The membrane was incubated for 1 h with HRP-conjugated secondary antibodies after being washed three times with TBST. The blots were visualized using imaging equipment (Bio-Rad). A loading control was used to standardize the expression levels of the target protein. ImageJ (image processing and analysis in Java, National Institutes of Health) was used to examine the gray values of each band.
Immunofluorescence
Prior to culturing the neurons, the culture dishes were pre-coated with slides of a suitable size. The culture medium was withdrawn immediately after sevoflurane or control treatment, and the vessels were rinsed three times with cold PBS. Each well was fixed for 30 min with 1 mL 4% paraformaldehyde, washed five times, and treated for 30 min with 0.3% Triton. Primary antibodies for AT8 (Cat. No. MN1020, 1:1,000; Thermo Fisher Scientific) and MAP2 (Cat. No. EPR19691, 1:1,000; Abcam) were incubated overnight in each well at 4°C. The slides were then cleaned three times and treated with secondary antibodies at room temperature for 1 h in the dark. Alexa Fluor® 594 goat anti-rabbit IgG (Cat. A-11012, 1:500; Thermo Fisher Scientific), and Alexa Fluor® 488 goat anti-mouse IgG (Cat. A-11001, 1:500; Thermo Fisher Scientific) were used as secondary antibodies. After being washed three times with PBS, the slides were incubated for 5 min at room temperature with DAPI (Cat. No. 104139; Abcam). The slides were then washed three times for 10 min using 0.01 M PBS. After treatment with anti-fluorescence quenching agent, the neurons were visualized using a fluorescence microscope and photomicrographs were captured.
Enzyme-linked immunosorbent assay (ELISA)
Mouse ELISA kits (TNF-α: Cat. No. E-EL-M0049c; IL-6: Cat. No. E-EL-M0044c, IL-1β: Cat. No. E-EL-M0037c; all from Elabscience, USA) were used to determine the expression levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) in each group. Briefly, 100 μL of the standard or sample was added to each well and incubated for 90 min at 37°C. Then, 100 μL of the specific antibody solution was added to each well and incubated for 60 min at 37°C. Each well was supplemented with 100 μL enzyme conjugate and incubated at 37°C for 30 min. After five washes, 90 μL substrate solution was added. Fifteen minutes later, 50 μL terminating solution was added to terminate the reaction. Finally, the optical density of each well was measured at 450 nm using a fluorescence reader and corrected at 570 nm.
Statistical analysis
Data are reported as mean ± standard deviation. For qPCR testing, there were four samples per group; in western blot tests, there were four samples per group; in ELISA tests, there were four samples per group; and in immunohistochemistry, there were three samples per group. The differences between the two groups in terms of biochemical data were evaluated using either the unpaired t-test (if values were normally distributed) or the Mann-Whitney test (if values were not normally distributed). Statistical significance was set at p < 0.05. GraphPad Prism software (version 5.0, GraphPad Software, USA) and SPSS (version 21.0, IBM, USA) were used for all statistical analyses.
Results
Sevoflurane anesthesia-induced ApoE mRNA, full-length ApoE, and 18-kDa ApoE fragments increased in hippocampal neurons extracted from WT but not ApoE-KO mice in vitro
We first compared the cell viability in different groups. We found that sevoflurane treatment could decreased the cell viability in both WT and ApoE-KO neuron (p < 0.05, WT + control group; p < 0.05, ApoE-KO + control group). We then compared the mRNA and protein levels of ApoE in hippocampal neurons extracted from WT and ApoE-KO mice following sevoflurane and control treatments. We found that the neurons expressed very tiny amounts of ApoE mRNA and full-length ApoE and did not produce any 18-kDa ApoE fragments under the control condition, whereas sevoflurane anesthesia significantly enhanced ApoE mRNA, total ApoE, full-length ApoE, and ApoE fragment levels (p < 0.05, WT + control group), and there was no ApoE mRNA, full-length ApoE, or 18-kDa ApoE fragment expression in ApoE-KO neurons (Figure 1). Differences in ApoE mRNA, full-length ApoE, and 18-kDa ApoE fragment expression between WT and ApoE-KO neurons. Neurons extracted (on the 5th day) from the hippocampi of WT and ApoE-KO mice were treated with 21% O2, 5% CO2, and 4.1% sevoflurane or 21% O2 and 5% CO2 for 4 h at 37°C. The neurons were harvested immediately after sevoflurane or control treatment. (a) Differences in cell viability in hippocampal neurons from WT and ApoE-KO mice following sevoflurane or control treatment (n = 4 samples per group). (b) ApoE mRNA detected by qPCR; fold change refers to WT + sevoflurane levels for comparison with experimental conditions (n = 4 samples per group). (c) Differences in full-length ApoE and 18-kDa ApoE fragment levels in hippocampal neurons from WT and ApoE-KO mice following sevoflurane or control treatment. (d, e) Summary of neuronal full-length ApoE and 18-kDa ApoE fragment protein levels; 100% of changes refer to WT + sevoflurane levels for comparison with experimental conditions (n = 4 samples per group). *p < 0.05, WT + control group.
ApoE-KO could not alleviate the tau phosphorylation and proinflammatory enhancement induced by sevoflurane anesthesia in hippocampal neurons in vitro
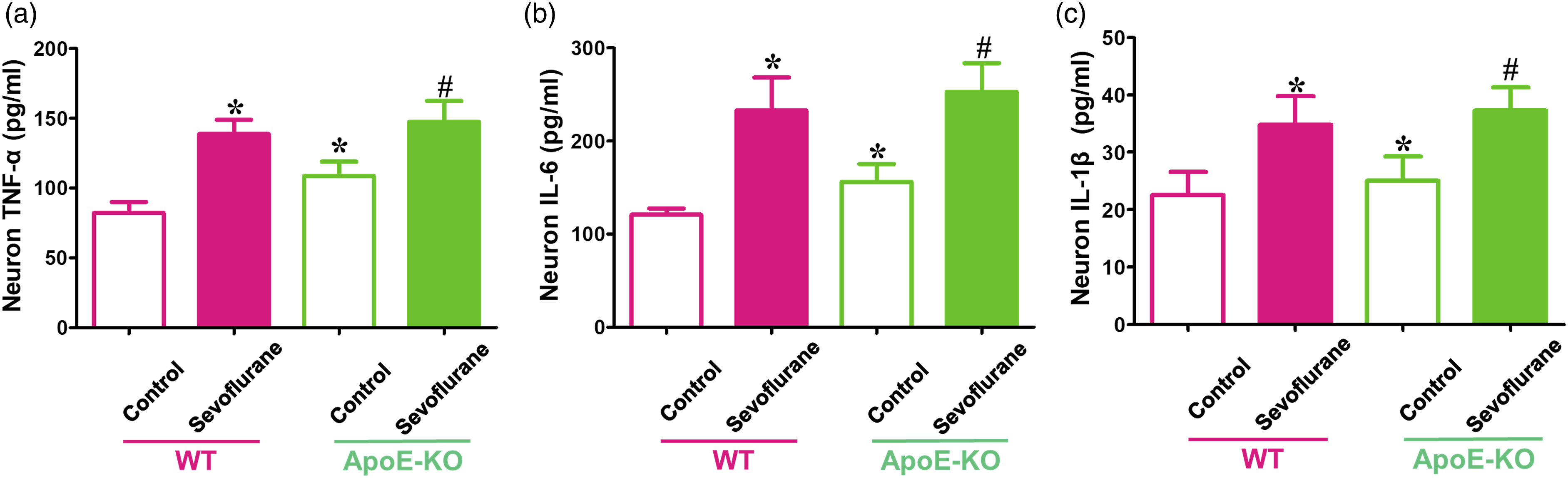
To examine whether total ApoE plays a critical role in the presence and severity of tau phosphorylation and neuroinflammation induced by sevoflurane anesthesia, hippocampal neurons extracted from WT and ApoE-KO mice were used in this study. We found that WT neurons treated with sevoflurane expressed higher levels of AT8, PHF1, and proinflammatory factors (TNF-α, IL-6, and IL-1β) than those in the control environment (p < 0.05, WT + control group) (Figures 2 and 3). However, the higher levels of phosphorylated tau protein and proinflammatory marker expression following sevoflurane anesthesia in WT neurons could not be reversed in ApoE-KO neurons (p > 0.05 for AT8 and PHF1 expression, ApoE-KO + control group; p < 0.05 for TNF-α, IL-6, and IL-1β levels, ApoE-KO + control group) (Figures 2 and 3). In control groups, ApoE-KO neurons expressed more TNF-α, IL-6, and IL-1β levels than WT neurons (p < 0.05, WT + control group) (Figure 2). Moreover, the immunofluorescence results also showed that there was more AT8-positive cell expression in sevoflurane-treated WT neurons and both control and sevoflurane-treated ApoE-KO neurons than in WT neurons under control conditions (p < 0.05, WT + control group) (Figure 2). These data illustrate that the lack of total ApoE expression in neurons could not prevent tau phosphorylation and neuroinflammation induced by sevoflurane anesthesia. Effects of sevoflurane on tau phosphorylation levels in primary hippocampal neurons from WT and ApoE-KO mice. Neurons cultured for 5 days were harvested immediately after sevoflurane or control treatment. (a) Western blot analyses of AT8 and PHF1 in neurons of WT and ApoE mice following sevoflurane or control treatment. (b, c) Summary of AT8 and PHF1 levels in different groups; 100% of changes refers to WT + control levels for comparison with experimental conditions (n = 6 samples per group). (d Immunostaining of AT8 (green fluorescence) and MAP2 (red fluorescence) expression in the neurons of WT and ApoE-KO mice following sevoflurane or control treatment; the positive cells that simultaneously expressed AT8 and MAP2 are shown by yellow fluorescence (white arrows). (e) Quantification of the ratio of positive cell (AT8/MAP2) expression (n = 3 samples per group). *p < 0.05, WT + control group. Effects of sevoflurane on proinflammatory factor expression in primary hippocampal neurons from WT and ApoE-KO mice. Neurons cultured for 5 days were harvested immediately after sevoflurane or control treatment. ELISA determination of (a) TNF-α, (b) IL-6, and (c) IL-1β levels in WT and ApoE-KO neurons following sevoflurane or control treatment (n = 4 samples per group). *p < 0.05, WT + control group; #p < 0.05 ApoE-KO + control group.
Differences in ApoE mRNA, full-length ApoE, and 18-kDa ApoE fragment expression between hippocampal neurons extracted from ApoE ε3 and ApoE ε2 mice in vitro
Because of the amino acid differences at position 158, ApoE ε2, which contains Cys158, is more stable than ApoE ε3, which contains Arg158.14,18 Therefore, compared with ApoE ε2, ApoE ε3 is more easily generated under certain conditions. The data showed that sevoflurane anesthesia decreased cell viability in ApoE ε3 neuron, but not in ApoE ε2 neuron (p < 0.05, ApoE ε3 + control group; p > 0.05, ApoE ε2 + control group). And sevoflurane increased ApoE mRNA and full-length ApoE levels compared with control treatment in both ApoE ε3 and ApoE ε2 neurons (p < 0.05, ApoE ε3 + control group; p < 0.05, ApoE ε2 + control group). In addition, there were obvious differences in 18-kDa ApoE fragment levels between sevoflurane and control treatment in ApoE ε3 neurons (p < 0.05, ApoE ε3 + control group), but there was no 18-kDa ApoE fragment expression in either the sevoflurane or control group of ApoE ε2 neurons (Figure 4). Differences in ApoE mRNA, full-length ApoE, and 18-kDa ApoE fragment expression between ApoE ε3 and ApoE ε2 neurons. Neurons extracted (on the 5th day) from the hippocampus of ApoE ε3-and ApoE ε2-targeted replacement mice were treated with 21% O2, 5% CO2, and 4.1% sevoflurane or with 21% O2 and 5% CO2 for 4 h in a 37-°C incubator. The neurons were harvested immediately after sevoflurane or control treatment. (a) Differences in cell viability in hippocampal neurons from ApoE ε3-and ApoE ε2-targeted replacement mice following sevoflurane or control treatment (n = 4 samples per group). (b) ApoE mRNA detected by qPCR; fold changes refer to ApoE ε3 + sevoflurane levels for comparison with experimental conditions (n = 4 samples per group). (c) Differences in full-length ApoE and 18-kDa ApoE fragment levels in hippocampal neurons from ApoE ε3-and ApoE ε2-targeted replacement mice following sevoflurane or control treatment. (d, e) Summary of neuronal full-length ApoE and 18-kDa ApoE fragment protein levels; 100% of changes refer to ApoE ε3 + sevoflurane levels for comparison with experimental conditions (n = 4 samples per group). *p < 0.05, ApoE ε3 + control group.
Sevoflurane-induced tau phosphorylation and neuroinflammation in neurons from ApoE ε3 mice could be reversed in neurons from ApoE ε2 mice in vitro
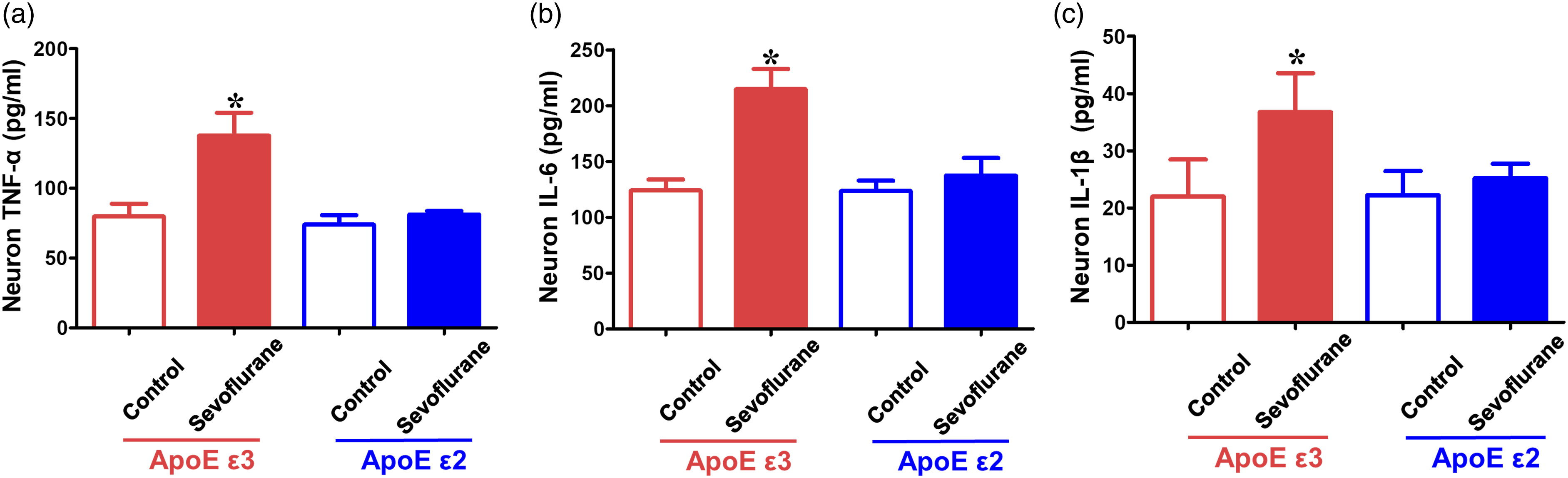
Neurons extracted from the hippocampi of ApoE ε3 and ApoE ε2 mice were used to detect whether the generation of 18-kDa ApoE fragments could affect tau phosphorylation and neuroinflammation caused by sevoflurane treatment in vitro. The results showed that ApoE ε3 neurons, but not ApoE ε2 neurons, expressed higher AT8, PHF1, TNF-α, IL-6, and IL-1β levels in the sevoflurane group than in the control group (p < 0.05, ApoE ε3 + control group; p > 0.05, ApoE ε2 + control group) (Figures 5 and 6). The 18-kDa ApoE fragments produced by ApoE ε3 neurons might be the main reason for the enhanced tau phosphorylation and neuroinflammation induced by sevoflurane anesthesia in vitro. Effects of sevoflurane on tau phosphorylation levels in primary hippocampal neurons from ApoE ε3-and ApoE ε2-targeted replacement mice. Neurons cultured for 5 days were harvested immediately after sevoflurane or control treatment. (a) Western blot analysis of AT8 and PHF1 in the neurons of ApoE ε3-and ApoE ε2-targeted replacement mice following sevoflurane or control treatment. (b, c) Summary of AT8 and PHF1 levels in different groups, 100% of changes refers to ApoE ε3 + control levels for comparison with experimental conditions (n = 6 samples per group). (d) Immunostaining of AT8 (green fluorescence) and MAP2 (red fluorescence) expression in the neurons of ApoE ε3-and ApoE ε2-targeted replacement mice following sevoflurane or control treatment; positive cells that simultaneously expressed AT8 and MAP2 are shown by yellow fluorescence (white arrows). (e) Quantification of the ratio of positive cell (AT8/MAP2) expression (n = 3 samples per group). *p < 0.05, ApoE ε3+ control group. Effects of sevoflurane on proinflammatory factor expression in primary hippocampal neurons from ApoE ε3-and ApoE ε2-targeted replacement mice. Neurons cultured for 5 days were harvested immediately after sevoflurane or control treatment. ELISA determination of (a) TNF-α, (b) IL-6, and (c) IL-1β levels in ApoE ε3 and ApoE ε2 neurons following sevoflurane or control treatment (n = 4 samples per group). *p < 0.05, ApoE ε3 + control group.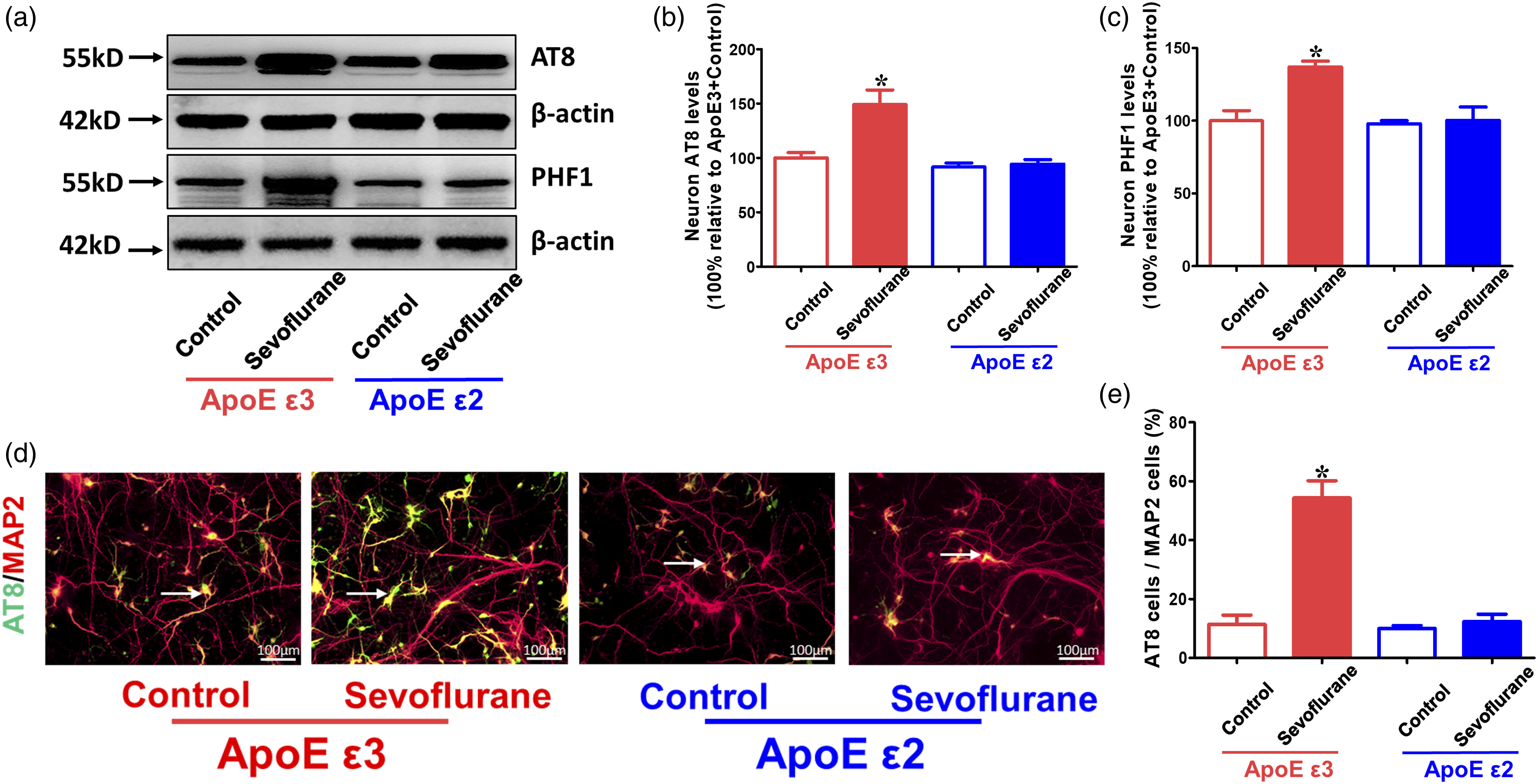
Discussion
In the present study, we found that sevoflurane anesthesia produced higher levels of ApoE mRNA, full-length ApoE, 18-kDa ApoE fragments, phosphorylated tau proteins (AT8 and PHF1), and proinflammatory factors (TNF-α, IL-6, and IL-1β) in WT neurons than in control neurons. However, ApoE deficiency did not attenuate sevoflurane-induced tau phosphorylation or neuroinflammation in ApoE-KO neurons. In contrast, sevoflurane administration induced tau phosphorylation and neuroinflammation in ApoE-KO neurons. Finally, lack of 18-kDa ApoE fragments, other than full-length ApoE, expressed in ApoE ε2 neurons attenuated sevoflurane-induced tau phosphorylation and neuroinflammation compared with ApoE ε3 neurons. These data illustrate that an increase in neurotoxic 18-kDa ApoE fragments contributes to differences in tau phosphorylation and neuroinflammation, with or without 4.1% sevoflurane administration.
Several published articles have indicated that hyperphosphorylated tau proteins in the brain play important roles in anesthesia-induced neurotoxicity and cognitive impairment in young mice1,7,21; however, the underlying mechanisms, especially the upstream mechanism by which anesthesia-induced tau phosphorylation leads to cognitive impairment in young mice, remain largely unknown. ApoE, a 35-kDa glycoprotein, is widely expressed in the human body and functions as a lipid transporter. 11 However, several emerging studies have shown that these functions may extend beyond lipid metabolism to influence AD or other neurodegenerative diseases including taupathy.12,22 ApoE has been reported to directly bind tau protein independently of Aβ in vitro, and neuronal expression of ApoE in vivo results in tau hyperphosphorylation.12,23,24 Previous studies found that sevoflurane anesthesia increased levels of ApoE and tau phosphorylation (AT8 and PHF1) as well as proinflammatory markers in neurons and hippocampus from the developing brain,10,25 which suggests that sevoflurane anesthesia-induced tau phosphorylation causes neuroinflammation and cognitive impairment might be related to the increase in ApoE.
Apolipoprotein E is a major carrier of cholesterol that is required for neuronal activity and injury repair in the brain; loss of ApoE might cause blood-brain barrier impairment and neuronal oxidative damage and limit synapse development, resulting in neurodegeneration.15,26,27 Thus, the generation of ApoE in neurons may be a neuroprotective function in the CNS. To further examine whether total ApoE plays a key role in the presence and severity of tau phosphorylation and neuroinflammation with sevoflurane anesthesia, WT and ApoE-KO neurons were used in the present study. We found that neurons from WT mice generated a small amount of full-length ApoE, and there was no ApoE fragment production in the control condition. Sevoflurane treatment enhanced both full-length and fragmented ApoE levels in WT neurons. Moreover, the neurons extracted from ApoE-KO mice expressed more phosphorylated tau proteins and proinflammatory markers in the control group than those from WT mice. These data indicate that when neurons receive dangerous stimuli, such as anesthesia, the ApoE produced by neurons may have protective effects against neuronal damage.
Apolipoprotein E is broken down by proteins in the human brain to produce truncated fragments, depending on the stability of different genotypes (ApoE ε2 > ApoE ε3 > ApoE ε4). 16 Although astrocytes highly express ApoE, there are no ApoE fragments when ApoE is overexpressed in astrocytes; however, in neurons, fragments are present under certain conditions. 24 Previous studies have reported that tau hyperphosphorylation, formation of Aβ plaques and NFTs, and the onset of neurodegeneration are directly associated with specific ApoE fragments.22,24,28 In the current study, we determined ApoE ε3 as the “wild type” allele because it is the most common subtype within the population, with an allele frequency of 0.7–0.8, and compared with the ε4 allele, fewer toxic fragments are produced under normal conditions.13,19 ApoE ε2, which is the most stable and least often split into fragments of all three alleles, was chosen to represent the “ApoE fragment deficiency” model.14,18 We found that sevoflurane anesthesia could induce higher full-length ApoE levels than the control condition in neurons extracted from ApoE ε3 and ApoE ε2 mice. However, sevoflurane treatment increased the toxic 18-kDa fragments levels only in ApoE ε3 mice, and no toxic 18-kDa fragments were produced in ApoE ε2 mice under either the anesthesia or control condition. In addition, the sevoflurane-induced phosphorylated tau enhancement and neuroinflammation in ApoE ε3 neurons could be mitigated in ApoE ε2 neurons. These data indicate that toxic 18-kDa ApoE fragments, but not full-length ApoE, might be the main reason for tau phosphorylation and neuroinflammation induced by sevoflurane anesthesia in neurons.
Conclusion
Full-length ApoE is protective and the 18-kDa fragment is toxic in neurons. Under normal conditions, ApoE, which is mostly full-length, is mainly produced by astrocytes and not neurons. However, when harmful stimuli exist, neurons may first generate full-length ApoE to activate and repair themselves after injury. Nevertheless, when harmful stimuli persist, the ApoE in neurons breaks into many fragments and disrupts the ApoE balance in the brain, which may lead to neuroinflammation and neurodegeneration, resulting in tau hyperphosphorylation in the brain and neurocognitive impairment. Overall, increasing the levels of 18-kDa ApoE fragments, but not full-length ApoE, could represent one of the underlying mechanisms of sevoflurane-induced tau phosphorylation and neuroinflammation in neurons.
Footnotes
Authors’ contributions
The study was planned and designed by Yang Yu, Man Yang, and Yonghao Yu. The experiment was conducted by Man Yang, Xiaoli Zhuang, Jiacheng Pan, Jiafeng Yu and Jingyu Feng. The manuscript was written by Yang Yu and Man Yang. Yonghao Yu edited and evaluated the manuscript. The final manuscript was reviewed and approved by all authors.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (grant nos. 82072150 and 82001149) and Tianjin Health Science and Technology Projects (grant no. KJ20023), the Science and Technology Development Fund of the Tianjin Education Commission for Higher Education (grant no. 2019KJ201), and the Tianjin Natural Science Foundation (grant no. 20JCQNJC01050).