
Abstract
Background
Gelsenicine, one of the most toxic alkaloids of Gelsemium elegans Benth (G. elegans), causes severe respiratory depression. However, its toxicity mechanisms are yet to be elucidated and no effective antidotes are available.
Objective
This study aimed to analyse the toxicity characteristics of gelsenicine.
Methods
Both acute and sub-acute toxicities were evaluated. Gelsenicine distribution and elimination in the central nervous system (CNS) and blood were observed. Effective antidotes for gelsenicine poisoning were screened.
Results
In the acute toxicity study, gelsenicine was highly toxic, and female rats exhibited greater sensitivity to gelsenicine than male rats (LD50 0.520 mg/kg vs 0.996 mg/kg, respectively). Death was primarily caused by respiratory failure. However, in the sub-acute toxicity study, no significant organ damage was observed. Gelsenicine was easily absorbed from the gastrointestinal tract and penetrated the blood–brain barrier, reaching peak concentrations in the CNS within 15 min and rapidly decreasing thereafter. Flumazenil or diazepam combined with epinephrine reversed gelsenicine toxicity and significantly improved survival rate in mice.
Conclusions
Gelsenicine is a highly toxic substance that affects nerve conduction without causing damage; the potential toxic mechanism is possibly associated with GABAA receptors. Our findings provide insights into the clinical treatment of gelsenicine-related poisoning and its toxicity mechanisms.
Introduction
Gelsemium elegans Benth is a traditional herbal medicine from Southeast Asia 1 with numerous biological activities, such as antitumour, 2 analgesic,3,4 anxiolytic 5 and anti-inflammatory activities. 6 However, G. elegans is highly toxic with a narrow therapeutic window, and clinical studies related to it are greatly limited.7,8 Cases of unintentional poisoning, food contamination, and suicides related to G. elegans are still being reported.9–11 Gelsenicine is a gelsedine-type indole alkaloid of G. elegans. 12 Though gelsenicine exerts biological effects, including analgesic, anxiolytic and sedative effects, on the central nervous system (CNS), it is the most toxic alkaloid of G. elegans. 13 Toxicological studies have shown that gelsenicine primarily affects the CNS, strongly inhibiting the respiratory centre, triggering respiratory failure and sometimes leading to death. 10 Analysis of the tissue distribution of gelsenicine revealed that it passes the blood–brain barrier and is detected in the CNS11,14; however, the transit mechanism is unclear. Meanwhile, gelsenicine toxicity differs with species; humans and rats are more sensitive than pigs and goats. This is related to the degree of absorption and metabolic breakdown of gelsenicine. 15 The main detoxification pathway is demethylation, 16 which is catalysed by cytochrome P450 monooxygenase CYP3A4/5.11,12 Studies on the detoxification of gelsenicine has shown that Mussaenda pubescens reduces the toxicity of gelsenicine by increasing the efflux of G. elegans alkaloids in Caco-2 cell monolayer models. 17 Pre-injection of flumazenil improved the survival rate of 14-(R)-hydroxy-gelsenicine-poisoned mice. 11 However, no effective treatment protocols have been reported for the clinical management of gelsenicine-related poisoning.
Studies on the acute toxicity of gelsenicine poisoning have been reported, but the consequences of low-level, chronic use of gelsenicine and the toxicity mechanism and targets are largely unknown. Besides, there are no effective antidotes available currently for clinical use. This study investigated the safety and toxicity characteristics of gelsenicine in rodents. The acute and sub-acute toxicity of gelsenicine was evaluated and its distribution and elimination in the blood and CNS were studied. Finally, potential antidotes were screened out after gelsenicine poisoning. This work may serve as a basis for further research on the mechanisms of toxicity and antidote regimens for gelsenicine.
Methods
Chemicals
Gelsenicine (C19H22N2O3, purity ≥ 99%) was purchased from Chengdu Must Bio Technology Co., Ltd. (Chengdu, China). Methanol, acetonitrile, and formic acid (all of chromatographic grade) were purchased from Merck (Darmstadt, Germany). Triple deionised water was purchased from Millipore (Billerica, MA, USA). All other chemicals used were of the highest grade and were obtained from reliable commercial sources.
Animals
Sprague–Dawley rats and ICR mice were used in this study which included 140 Sprague–Dawley rats (200 ± 20 g) and 300 ICR mice (20 ± 2 g) of both sexes and the corresponding diets were purchased from Hunan SJA Laboratory Animals Co., Ltd. (Changsha, China). All animal experiments were conducted in a specific pathogen-free laboratory animal room (temperature 22 ± 2°C, humidity 60%, and 12 hrs light/dark cycles); animals were allowed to acclimatise for one week before the initiation of the experiments and provided free access to food and water. Animals were randomly assigned (per weight) to each group. All experiments were carried out in compliance with the Guide for Care and Use of Laboratory Animals
18
and approved by the Ethical Committee of Animal Care of the Hunan Anshengmei Drug Research Institute Co., Ltd. (Ethics No. ASM-2019031). The experimental design of the animals in this is shown in Figure 1. Flowchart depicting the study design.
Acute toxicity study of gelsenicine
The oral acute toxicity study was carried out according to the Economic Cooperation and Development (OECD) guidelines No. 423. 19 Considering that gelsenicine showed a significant sex-based difference in the preliminary experiments, the lethal dose 50 (LD50) of gelsenicine in male and female rats was determined using five dose levels (0.6, 0.8, 1.1, 1.5, and 2.0 mg/kg in male and 0.34, 0.45, 0.6, 0.8, and 1.1 mg/kg in female, n = 5). The rats were deprived of food overnight before the oral administration. Gelsenicine was diluted in normal saline at the required dose concentration (10 mL/kg) then gavage. After dosing, clinical signs, time of poisoning, and death were observed and recorded. The experiment lasted for 14 consecutive days after treatment, and the results were assessed using the Bliss method.
Sub-acute toxicity study of gelsenicine
Additionally, we exposed the rats to the maximum tolerated dose (MTD) of gelsenicine to investigate sub-acute toxicity. The rats were divided into four groups, that is, male-control, male-gelsenicine, female-control, and female-gelsenicine (n = 5). The male-control and female-control groups were administered normal saline, whereas male-gelsenicine and female-gelsenicine groups were administered gelsenicine at doses of 0.6 and 0.3 mg/kg, respectively. For oral toxicity studies, repeated doses were administered to rats according to the OECD guidelines No. 407 for testing of chemicals. 20 Oral gavage was performed daily for 28 consecutive days at the same time.
General observations and body weight
Abnormal clinical signs and behavioural changes were also assessed on a daily basis during the study period. The body weight was measured and recorded at the beginning and every three days thereafter.
Sample collection
At the end of the study, all animals were fasted overnight with water ad libitum. Blood samples were collected from the retro-orbital region of the rats for the measurement of biochemical and haematological parameters. After being sacrificed by cervical dislocation, organ coefficients and histopathology parameters were examined.
Biochemical parameters
Blood samples collected in non-treated tubes were allowed to coagulate for 45 min at room temperature and then centrifuged (2500 × g, 25°C) for 20 min to obtain the serum. All serum samples were evaluated using an automatic biochemical analyser FAITH-1000 (Laura, Nanjing, China) and an electrolyte analyser KH-996 (Kinghawk, Beijing, China).
Haematological parameters
Blood samples were collected in EDTAK2 anticoagulation tubes. The analysis was performed using an automated blood cell analyser DF50 (Dymind Biotech, Shenzhen, China).
Weight and histology of organs
All rats in the study were subjected to detailed gross necropsy in the anatomy process. The organs were removed immediately after euthanasia by cervical dislocation and weighed to calculate the relative organ weight (organ weight/body weight, %). The heart, liver, spleen, lungs, kidneys, spinal cord, and brain were then placed in 10% neutral-buffered formalin overnight, processed for paraffin embedding, sliced into standard thin sections, and stained with haematoxylin-eosin, and then examined via microscopy.
Distribution and elimination of gelsenicine in the CNS
Thirty male and thirty female rats were divided into six groups. All rats were administered 0.3 mg/kg body weight of gelsenicine by gavage. Rats were sacrificed 5, 10, 20, 40, 60, and 90 min (n = 5) after gavage. The brain and spinal cord were removed quickly, and the spinal cord, cerebellum, brainstem, cerebral cortex, hippocampus, and striatum were dissected. The tissue samples were weighed and collected in 2.0 mL centrifuge tubes. One millilitre of cold water/1.0 g of tissue was added, and the tissues were homogenised for 2 min in a high-speed frozen grinder before storage at −80°C until analysis.
For pharmacokinetic studies, blood samples from five male and five female rats were collected from the orbital vein at 0.08, 0.17, 0.33, 0.67, 1, 1.5, 2, 4, 6, 8, 12, and 24 hrs. After centrifuging (2500 × g, 20 min, 4°C), the plasma samples were collected and stored at −80°C until further analysis.
Gelsenicine levels were quantified in the serum and tissue samples using the ultra-performance liquid chromatography coupled with tandem mass spectrometry (UPLC-MS/MS) method established and published by our group.15,21–23 Standard stock solution of gelsenicine (1.0 mg/mL) was prepared in pure methanol. Next, a working standard solution of gelsenicine was prepared at the concentrations of 0.1, 0.2, 0.5, 1.0, 2.0, 5.0, 10.0, 20.0 and 50.0 μg/L, with rat plasma used as a blank. 23 Notably, the methods were optimised with respect to sample extraction, limits of quantitation (LOQ), and matrix-matched standard curves in stages according to the guidelines of the European Medicines Agency. The limit of detection (LOD) of the optimised method was 0.1 μg/L. All tissue homogenates were treated in accordance with the optimised method, and finally, 10.0 μL samples were loaded onto the UPLC-MS/MS system for analysis.
Screening of effective antidotes for gelsenicine poisoning
Candidate drug information
All candidate drugs used in this study were obtained from reliable commercial sources; epinephrine (1 mg/mL) from Harbin Pharmaceutical Group, Sanjing Pharmaceutical Co., Ltd. (Harbin, China); flumazenil (0.1 mg/mL) from Hunan Zhengqing Pharmaceutical Group Co., Ltd. (Changsha, China); diazepam (5 mg/mL) from Tianjin King York Pharmaceutical Co., Ltd. (Tianjin, China); strychnine (2 mg/mL) from Harbin Sanma Veterinary Medicine Co., Ltd. (Harbin, China); naloxone (0.4 mg/branch) from Cisen Pharmaceutical Co., Ltd. (Jining, China); nikethamide (250 mg/mL) from Shanghai Harvest Pharmaceutical Co., Ltd. (Shanghai, China); lobeline (3 mg/mL) from Beijing Yookon Pharmaceutical Co., Ltd. (Beijing, China); and neostigmine (0.5 mg/mL) from Shanghai Sine Pharmaceutical Group Co., Ltd. (Shanghai, China).
Screening for effective gelsenicine antidotes
An acute poisoning model was needed to screen for effective gelsenicine antidotes. On account of animal ethical requirements, small size and toxicity-sensitive ICR mice were selected as the experimental model. The selected dose of gelsenicine was 1.5 mg/kg based on the pre-experiment LD50 of gelsenicine in mice by oral gavage which led to the death of approximately 70–80% of the mice and caused poisoning symptoms in 100% of them. ICR mice were used to simulate intoxication and divided into one control group and eight drug-treated groups (n = 20, half males and half females). After mice were administered with gelsenicine, the drug-treated groups were intraperitoneally injected with the candidate drugs. The eight different drugs were flumazenil, diazepam, epinephrine, strychnine, naloxone, nikethamide, lobeline and neostigmine. The dosages of the drug candidates were calculated based on the body surface area conversion in accordance with guidelines for the correlation of the dose equivalents between humans and laboratory animals: flumazenil (0.2 mg/kg), diazepam (5 mg/kg), epinephrine (0.2 mg/kg), strychnine (0.3 mg/mL), naloxone (0.5 mg/kg), nikethamide (100 mg/kg), lobeline (2 mg/kg), and neostigmine (0.13 mg/kg). The time of intoxication and death was recorded after gavage; head tremors were used as an orientation guide. Survival curves were used to calculate whether the drug could relieve poisoning symptoms, delay the time of death, or reduce the number of deaths.
Optimisation of the detoxification effect
Combinations of the effective drugs were used to optimise the detoxification effect of gelsenicine poisoning. ICR mice were divided into one control group and five drug-treated groups (n = 20, half males and half females). All mice were orally administered gelsenicine (1.5 mg/kg). The five drug treatment groups were intra-peritoneally injected with flumazenil, diazepam, epinephrine, flumazenil + epinephrine, and diazepam + epinephrine, after gavage. The dosages of drugs, monitoring index, and data analysis methods were identical to those described in 2.6.2.
Statistical analysis
Data were presented as mean ± standard deviation and analysed using GraphPad Prism 7 (GraphPad Software, LaJolla, USA). Student’s t-test and one-way analysis of variance were used for statistical analyses. Survival curves were assessed using the log-rank test. Additionally, the UPLC/MS/MS data were analysed using Analyst 1.6.3 software (AB SCIEX, Framingham, MA, USA). The LOD and lower LOQ values were obtained via direct observation of the signal-to-noise ratio on the total ion chromatograms graphs. Finally, all analyses of toxicokinetic parameters were conducted using Winnonlin trials (Pharsight, NC, USA) for non-compartmental model analysis.
Results
Acute toxicity study
The poisoning symptoms, dose-effect, poisoning time, and death time were analysed based on acute toxicity (Figure 2). Gelsenicine-induced death in rats in a dose-dependent manner. The early toxic effects observed were decreased spontaneous activity, tachypnoea, prominent protrusion of the eyeballs, and head tremors. Therefore, we used head tremors to define the time of poisoning (Figures 2(a) and (b)). The incubation period was very short, with the onset of symptoms typically appearing 10 min after ingestion; symptoms of dyspnoea, such as abdominal breathing, open-mouth-breathing, cyanosis, and convulsions, were observed. Death was primarily triggered by respiratory failure; the rats that did not die within 1 hr survived throughout the entire observation period (Figures 2(c) and (d)). The time to death was typically within 40 min after ingestion (approximately 20 min in most cases; Figure 2(e)). Interestingly, a significant gender difference was observed for the LD50 of gelsenicine; 0.996 and 0.520 mg/kg for male and female rats, respectively (Figure 2(f)). According to the standard recommended by the World Health Organisation for classifying the acute toxicity of compounds by oral administration,
24
our results suggest that gelsenicine is a highly toxic substance. Of note, necropsies were performed after poisoning-induced death and no obvious pathological changes in the main organs, except extravasated blood in the lungs, liver, and right heart compartments, were observed. Acute toxicity of gelsenicine in male and female rats (n = 5 per group). (a, b) The asymptomatic percentage curve in male and female rats. (c, d) The survival percentage curve in male and female rats. (e) The mean time of poisoning and death in female and male rats. (f) The LD50 of gelsenicine in female and male rats.
Sub-acute toxicity study
General observations
The MTD of gelsenicine was administered to rats daily, for 28 consecutive days. During the study period, none of the animals treated with gelsenicine died. After administration, most animals displayed toxic symptoms, including decreased activity, closed eyes, head tremors, and abdominal breathing but recovered later.
Body weight
The body weight gain in the gelsenicine groups was lower than that in the control group (Figure 3). Twenty-eight days after initiating gelsenicine administration, the body weight of males and females treated with gelsenicine was reduced by 10.8% (p < .01) and 4.2% versus the control, respectively. Gelsenicine may have caused a reduction in rat body weight. After gelsenicine administration, rat activity declined significantly, which in turn affected food intake and resulted in weight loss. Effects of the oral administration of gelsenicine for 28 consecutive days on the mean body weight of rats (n = 5 per group). *p < .05, **p < .01 versus control.
Biochemical analysis
Biochemical parameters of gelsenicine-exposed rats for 28 consecutive days.

The values are expressed as the mean ± standard deviation (n = 5 per group). ALT, alanine aminotransferase; AST, aspartate aminotransferase; TP, total protein; ALB, albumin; TBIL, total bilirubin; ALP, alkaline phosphatase; BUN, blood urea nitrogen; CREA, creatinine; TG, triglyceride; CHO, cholesterol; GLU, glucose; CK, creatine kinase; A/G, albumin to globulin; TCa, total calcium. *p < .05, **p < .01.
Haematological analysis
Haematological parameters of rats exposed to gelsenicine for 28 consecutive days.

The values are expressed as the mean ± standard deviation (n = 5 per group). WBC, white blood cell count; RBC, red blood cell count; MCV, mean corpuscular volume; MCH, mean haemoglobin content; MCHC, mean haemoglobin concentration; MPV, mean platelet volume; PDW, platelet distribution width; PCT, thrombocytocrit. *p < .05, **p < .01.
Organ weight and histological analysis
Effect of oral administration of gelsenicine to rats for 28 consecutive days on the organ weight index.

The values are expressed as the mean ± standard deviation (n = 5 per group). The body weight is expressed in absolute (g) and relative (% body weight) units. *p < .05, **p < .01.

Histopathological examination of the vital organs after the administration of gelsenicine to rats for 28 days. H&E stained preparations are shown; original magnifications: liver × 100, other organs × 400 (n = 5 per group).
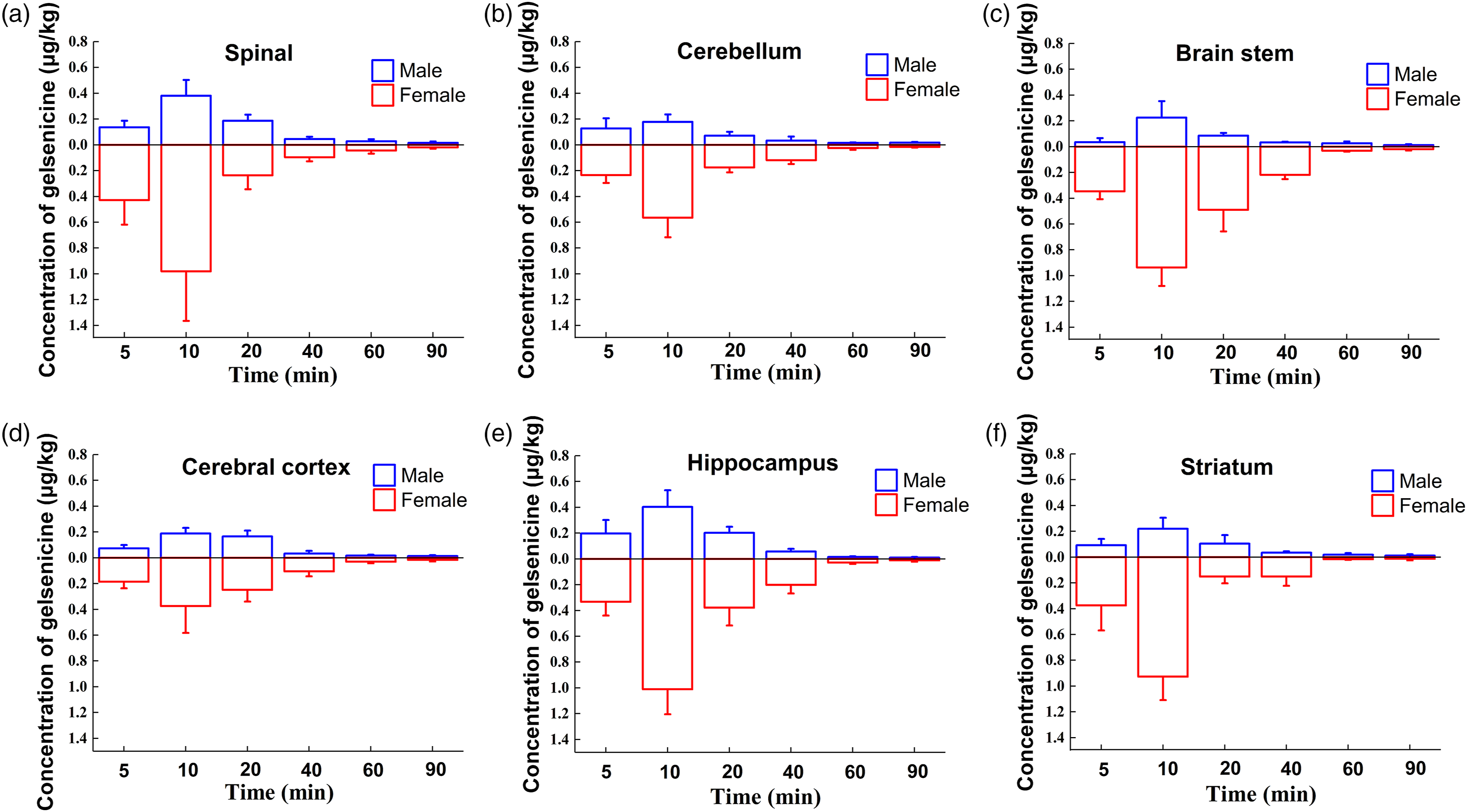
Distribution and elimination of gelsenicine in the CNS of rats
To comprehensively evaluate the toxic effect of gelsenicine, the distribution and elimination of gelsenicine in the CNS after a single oral administration were evaluated. Gelsenicine was rapidly absorbed from the gastrointestinal tract into the blood (Figure 5)and it easily penetrated the blood–brain barrier into the CNS (Figure 6). The plasma toxicokinetic drug-time curves and parameters showed that the plasma gelsenicine concentration of female rats was significantly higher than that of male rats at the same administered dose (Table 4); the maximum plasma concentration (Cmax) and area under the concentration-time curve values for female rats were 2.3 – and 3.6 – fold higher than those for male rats. However, the time to reach the peak concentration (Tmax) in females was 0.8 times that in males, and the half-elimination time (T1/2) in females was 3.3 times that in males, meaning that the absorption rate and absorption degree of gelsenicine in female rats were higher than those in male rats, whereas the elimination rate was lower in female rats than in male rats. This was consistent with the distribution and elimination of gelsenicine in each region of the CNS of rats. However, in the spinal cord, brainstem, hippocampus, and striatum, the concentrations were higher than those in other regions. Of note, gelsenicine reached peak concentration in the CNS at approximately 10 min after ingestion and was rapidly eliminated after 20 min; gelsenicine showed rapid absorption (Tmax < 13 min) and metabolism (T1/2 < 1.5 hr) rates. Toxicokinetics of gelsenicine in male and female rats after a single oral gavage. The average plasma concentration-time profile is shown (n = 5 per group). Gelsenicine distribution in various regions of the rat central nervous system after a single oral gavage (0.3 mg/kg body weight, n = 5 per group). (a) Spinal cord, (b) cerebellum, (c) brainstem, (d) cerebral cortex, (e) hippocampus, and (f) striatum. Comparisons of the concentration-time gelsenicine profiles in male versus female rats are presented. Plasma toxicokinetic parameters of rats after the single oral gavage administration of gelsenicine (0.3 mg/kg body weight) (n = 5 per group). The values are expressed as the mean ± standard deviation (n = 5 per group). Cmax, maximum plasma concentration; Tmax, time to peak plasma concentration; T1/2, half-life; AUC0-t, area under the concentration-time curve from time 0 to t; AUC0-∞, area under the concentration-time curve from time 0 to infinity; CL, total plasma clearance; MRT, mean residence time; Vd, apparent distribution volume.

Screening for effective gelsenicine antidotes
Preliminary screening
Considering the acute poisoning symptoms of gelsenicine, we recorded the time of poisoning and death in animals treated with different compounds for identifying an effective antidote; eight candidate drugs were screened (Figure 7): neostigmine (an acetylcholinesterase inhibitor), strychnine, flumazenil, lobeline, and naloxone (drugs that relieve respiration depression), epinephrine (a cardiotonic), and diazepam (a sedative-hypnotic). In ICR mice, the symptoms of gelsenicine poisoning appeared approximately 5 min after gavage, and acute death occurred in approximately 10 min (data not shown); The time of poisoning after the injection of epinephrine was delayed significantly from 5.4 to 9.7 min (Figures 7(a) and (c)). The mean survival time was also significantly longer in the epinephrine- and diazepam-treated groups; the time to death in the diazepam group was delayed from 11 to 28.8 min, which was more significant than that in the epinephrine group (from 11 to 15.7 min; p < .01; Figures 7(b) and (c)). Overall, among the eight compounds, only flumazenil, diazepam, and epinephrine increased the survival rate of mice (Figures 7(b) and (d)), from 25% to 50%, 65%, and 55%, respectively. Therefore, we selected these three drugs for subsequent studies. Screening of different candidate drugs for determining a gelsenicine-poisoning antidote. The times of poisoning (a) and death (b) were determined in the different groups based on the survival curves (gelsenicine dosage of 1.5 mg/kg; administered via gavage, n = 20 per group). (c) Quantitative data are also shown with the respective statistics. *p < .05, **p < .01 versus control. (d) The survival rate in each group was also determined.
Co-administration of drugs to optimise the detoxification effect
Considering that epinephrine is the first-line drug for cardiopulmonary resuscitation, increasing the force and rate of myocardial contraction and relaxing the bronchial smooth muscles resulting in bronchodilation,
25
we decided to use this drug in two different combinations for the optimisation of the detoxification effect (Figure 8). Co-administration by intra-peritoneal injection of diazepam and epinephrine was the most effective gelsenicine antidote, with no typical symptoms during the observation period (Figure 8(a)) with a 95% survival rate (Figure 8(b)). The co-administration of flumazenil and epinephrine also showed favourable results with a 65% asymptomatic rate (Figure 8(a)) and a survival rate of 85% (Figure 8(b)). Overall, these results suggest that the co-administration of diazepam and epinephrine has great detoxification potential and should be used to treat gelsenicine-induced acute toxicity. The co-administration of different drugs leads to better detoxification effects (n = 20 per group). (a) The asymptomatic rate in the different groups. (b) The survival rate in the different groups. FLM, flumazenil; DAP, diazepam; EPI, epinephrine. *p < .05, **p < 0.01 versus control.
Discussion
Refining basic toxicological data can play a key role in the risk assessment of poisons and the consequent avoidance of poisoning accidents. The safety and potential hazard of many chemicals have been well characterised and mitigated. 26 Gelsenicine is the most toxic component of the traditional herbal medicine G. elegans. The toxicity mechanism of gelsenicine has not yet been elucidated. Besides, there are no effective antidotes available for gelsenicine-induced poisoning. In this study, we evaluated the general toxicity characteristics of gelsenicine and determine effective antidotes.
The determination of acute toxicity allows the assessment of the toxic potential of a substance, 27 whereas that of sub-acute toxicity allows the disclosure of target organs after exposure to repeated doses. In this study, gelsenicine was defined as a very toxic substance with a short onset period and a low LD50 (less than 1 mg/kg via gavage administration). Curiously, female rats were more sensitive to gelsenicine than male rats. Sex-based gender toxicity may be related to cytochrome P450 enzymes; for instance, CYP3A4, which is important for the metabolic detoxification of gelsenicine, is expressed at significantly higher levels in female rats than in male rats.12,28 In the sub-acute toxicity experiments, body weight gain was lower in both male and female rats after receiving gelsenicine for 28 days. A comprehensive analysis of the biochemical and haematological parameters, vital organ index, and pathological sections showed an increase in ALT in both female and male rats and a decrease in the liver index in male rats. High levels of ALT were associated with liver damage from exogenous substances, 29 but the overall biochemical data and examination of liver pathological sections revealed that gelsenicine caused limited damage to the liver and no parenchymal damage. The concentration of Na+, Cl-, and TCa2+ in serum changes with statistically significance, but the variation range of data was small. We cannot say that the difference in electrolyte levels was abnormal in gelsenicine groups. Overall, in line with other reports on G. elegans, gelsenicine in this study triggered respiration failure 10 without gastrointestinal toxicity, hepatotoxicity, nephrotoxicity, and cardiotoxicity. 7 Therefore, these data suggested that gelsenicine is an acute neurotoxin that can lead to death; but it did not produce marked cumulative toxic effects and substantial organ damage in sub-acute toxicity.
To further investigate the toxic effects of gelsenicine, the concentration-dependent effect in the CNS was studied. The pharmacokinetic and tissue distribution experiments demonstrated that G. elegans alkaloids could effectively cross the blood-brain barrier in rats.11,14,30,31 Our results showed that gelsenicine was rapidly absorbed from the gastrointestinal tract and easily penetrated the blood-brain barrier, rapidly distributing throughout the CNS; however, it was also rapidly eliminated. These results are consistent with those reported in the context of different models; the peak concentration can be reached within half an hour, whereas the elimination half-life ranges from 1.37 32 to 5.0915 h. Notably, the concentration of gelsenicine in the CNS was closely correlated with the symptoms of toxicity. In fact, female rats exhibited superior bioavailability than male rats for the same gelsenicine dose, which may explain the low LD50 in female versus male rats exposed to gelsenicine. The diagnosis of gelsenicine poisoning is challenging and requires the timely collection of urinary samples. 8
Cases of G. elegans poisoning are due to the accidental ingestion (when confused with honeysuckle or contaminated food, honey). However, as G. elegans is used as a traditional folk medicine, medication errors may also lead to poisoning incidents; also, its use for suicide or homicide purposes cannot be excluded. 8 Still, there are no specific effective drugs for the management of gelsenicine-related poisoning; gastric lavage, activated charcoal, catharsis, or blood purification, which are the general approaches to speed up the elimination of poisons, are commonly used. Additionally, respiratory support (including invasive ventilation) is typically required. Acute neurotoxicity induced by gelsenicine has a quick onset, with the potential to rapidly evolve into respiratory depression, blood pressure reduction, and respiratory failure, which cannot be reversed. Respiratory depression is due to the CNS, and not the peripheral nervous system.33,34 Therefore, the above treatments are associated with low rescue efficiency. Based on previous findings, the pre-injection of flumazenil and picrotoxin for 15 min has a preventive effect on 14-(R)-hydroxy-gelsenicine poisoning. 11 Additionally, the pre-injection of pentobarbital or diazepam for 30 min significantly delays the onset of convulsions (with no impact on the survival rate) in G. elegans-poisoned rats. 33 Most studies have evaluated the prophylactic effect, not the detoxification potential. Here, we show that the administration of flumazenil, diazepam, and epinephrine can prevent the toxic effects of gelsenicine and increase the survival rate of poisoned mice. Strikingly, intra-peritoneal administration of epinephrine in combination with flumazenil or diazepam significantly prevented mortality and contributed to the rapid recovery of gelsenicine-poisoned mice; the combination of epinephrine with diazepam was the more effective alternative. In contrast, the administration of drugs, such as nikethamide, strychnine, naloxone, lobeline, and neostigmine, did not prevent/reverse gelsenicine-mediated toxicity.
Benzodiazepines are GABAA receptor ligands widely used for the treatment of epilepsy, anxiety, and insomnia.35–37 Diazepam, a classical benzodiazepine drug, is a positive allosteric modulator of GABAA receptors with a wide range of pharmacological effects, from sedation to anaesthesia. 38 In contrast, flumazenil is a GABAA receptor antagonist used clinically in the context of benzodiazepine overdoses or general anaesthesia reversal 39 ; it can compete for the α1-γ2 high-affinity benzodiazepine site of the receptor.36,40 Our results suggest that both diazepam and flumazenil are effective gelsenicine antidotes, although the former showed better detoxification effects than the latter. These findings were unexpected; however, they clearly support the notion that the mechanism behind gelsenicine toxicity is dependent on GABAA receptors and the impairment of neuronal activity in the CNS. The same hypothesis has been previously proposed in the context of 14-(R)-hydroxy-gelsenicine. 11 Additionally, epinephrine is a powerful vasopressor that increases the force and rate of myocardial contraction and relaxes bronchial smooth muscles, leading to bronchodilation.41,42 We believe that the administration of epinephrine counteracted the respiratory arrest and blood pressure reduction caused by gelsenicine. Therefore, epinephrine in combination with diazepam or flumazenil probably addresses gelsenicine poisoning on two different fronts: directly antagonising gelsenicine and indirectly alleviating the gelsenicine-derived symptoms.
However, further investigations, including patch-clamp assays are warranted to investigate the possible effects of gelsenicine on ion channels and neurotransmitter receptors in the CNS. Humans and rats are sensitive to gelsenicine toxicity. Our data support that diazepam and epinephrine are effective in the treatment of gelsenicine-related poisoning. Further independent studies are necessary to validate/replicate the results in this study and support the clinical application of gelsenicine-poisoning antidotes.
Conclusions
Altogether, our results suggest that gelsenicine toxicity is associated with the fast distribution and clearance of gelsenicine throughout the CNS, with no apparent tissue damage. Additionally, we show that flumazenil and diazepam have detoxification effects in the context of gelsenicine poisoning, particularly when used in combination with epinephrine. These results support the hypothesis that gelsenicine toxicity is associated with GABAA receptor-related respiratory depression. Our data support the clinical use of diazepam and epinephrine for the treatment of gelsenicine-related poisoning.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the National Key Research and Development Programme of China (2017YFD0501403), The National Natural Science Foundation of China (31972737), and Doctoral Scientific Research Foundation of Henan University of Animal Husbandry and Economy (M4030068).