
Abstract
Background: Mitochondria are the main target organelles through which drugs and chemicals exert their toxic effect on cardiomyocytes. The mitochondria-related mechanisms of celecoxib-induced cardiotoxicity have been extensively studied. Accumulated evidence shows natural molecules targeting mitochondria have proven to be effective in preventing cardiotoxicity. Purpose: In the present study, we examined the ameliorative effect of gallic acid (GA) against celecoxib-induced cellular and mitochondrial toxicity in isolated cardiomyocytes and mitochondria. Research Design: The isolated cardiomyocytes and mitochondria were divided into various group, namely, control, celecoxib, celecoxib + GA (10, 50, and 100 µM). Several cellular and mitochondrial parameters such as cell viability, lipid peroxidation, succinate dehydrogenase (SDH) activity, reactive oxygen species (ROS) formation, mitochondrial membrane potential (MMP) collapse, and mitochondrial swelling were assessed in isolated cardiomyocytes and mitochondria. Results: Our results showed that administration of celecoxib (16 µg/ml) induced cytotoxicity and mitochondrial dysfunction at 6 h and 1 h, respectively, which is associated with lipid peroxidation intact cardiomyocytes, mitochondrial ROS formation, MMP collapse, and mitochondrial swelling. The cardiomyocytes and mitochondria treated with celecoxib + GA (10, 50, and 100 µM) significantly and dose-dependently restore the altered levels of cellular and mitochondrial parameters. Conclusions: We concluded that GA through antioxidant potential and inhibition of mitochondrial permeability transition (MPT) pore exerted ameliorative role in celecoxib-induced toxicity in isolated cardiomyocytes and mitochondria. The data of the current study suggested that GA supplementation may reduce celecoxib-induced cellular and mitochondrial toxicity during exposure and may provide a potential prophylactic and defensive candidate for coxibs-induced mitochondrial dysfunction, oxidative stress, and cardiotoxicity.
Keywords
Introduction
Cardiac cells use a numerous amount of adenosine triphosphate (ATP) to perform their continuous activities. 1 Large amounts of this energy are provided by cardiac mitochondria. Therefore, to maintain constant ATP production, damaged mitochondria are rapidly replaced by newly synthetized organelles in cardiomyocytes. 1 Due to the importance of mitochondria in energy production and reactive oxygen species (ROS) in cardiomyocytes, drugs and chemicals that interfere with the mitochondrial function are expected to induce ATP depletion and oxidative stress in these cells. 1 Finally, depletion of ATP and ROS generation may lead to subsequent myocardial dysfunction. Drugs may directly and indirectly cause mitochondrial dysfunction through several potential ways such as inhibition of mitochondrial replication, interaction with the electron transport chain (ETC), resulting in reduction of mitochondria number, uncoupling of electron transport from ATP production and oxidative stress. 2 These mechanisms gradually reduce the function of mitochondria in various tissues specially in metabolically active organs such as the heart, resulting in cardiotoxicity. 1
Various clinical and experimental studies have been made regarding the safety and effectiveness of nonsteroidal anti-inflammatory drugs (NSAIDs) in cardiovascular diseases (CVDs) during the past few years. 3 The risk of myocardial infarction, atrial fibrillation, heart failure, and cardiotoxicity has been reported in patients after use of NSAIDs with a history of pathological and non-pathological conditions. 3 It has been reported that the use of NSAIDs increases the rate of cardiovascular events like myocardial infarction, stroke, and cardiovascular death. 4 Several studies suggest that coxibs such as rofecoxib and celecoxib elevate the rate of incidences of cardiovascular disorders compared to nonselective NSAIDs.5,6 Various mechanisms have been proposed to induce cardiac toxicity induced by NSAIDs. But among them, mitochondrial dysfunction and oxidative stress are common mechanisms of different NSAIDs. 3 These drugs have been shown to have toxic effects on the mitochondria resulting in the increased generation of ROS. 3 Previous studies suggest that mitochondrial complex III and IV are affected by NSAIDs resulting in increased ROS formation. 3 Celecoxib as selective COX 2 inhibitor dose-dependently induces mitochondria swelling, ROS formation, and cytochrome c release and exerts inhibitory effects on the ETC.7,8 Rat embryonic H9c2 cardiac cells treated to celecoxib demonstrated a decrease in cell viability, and this was associated with a decrease in the expression of Bcl2 protein. 9 Above studies suggest that mitochondria are the main target organelles through which celecoxib exerts its toxic effect on cardiac cells. Due to the large number of mitochondria in the heart and the high susceptible of these organelles to drug-induced oxidative stress, it is suggested that antioxidant compounds could reduce the mitochondrial toxicity caused by NSAIDs, especially celecoxib. 10
Gallic acid (GA), 3,4,5-trihydroxybenzoic acid, is a phenolic compound found in many plants possessing antioxidant activity. 11 This phenolic compound is the key component of cosmetics and different therapeutic herbal medicines. 12 Due to its potent property of scavenging ROS, like hydrogen peroxide (H2O2), superoxide anions, hypochlorous acid (HOCl), hydroxyl radicals (OH), and ameliorating oxidative stress, especially drug-induced oxidative stress, this compound has been used in various studies. 13 Already, pharmacological effects of GA such as the anti-bacterial, anti-diabetic, anti-angiogenic, anti-inflammatory, anti-cancer, and anti-oxidant have been reported. 14 Furthermore, the cardioprotective effect of GA has been established in several studies.15–18 Earlier study reveals that GA can provide protection against oxidative stress and mitochondrial dysfunction induced by bisphenol A. 19 It has been suggested that GA protects the mitochondria probably by reducing the oxidative stress. Also, GA protects the mitochondrial surface from bisphenol A–induced oxidative damages. 19 Due to the promising effect of GA against cardiotoxicity induced by drugs and chemicals, its protection effect against oxidative stress and mitochondrial toxicity, it deserves to be more studied. Therefore, here we purpose to study the effect of GA against celecoxib-induced toxicity in isolated cardiomyocytes and mitochondria obtained from rat heart.
Materials and methods
Chemicals
Fetal bovine serum (FBS), collagenase (Type II), 199 Medium, streptomycin and penicillin Solution, creatine, taurine, 2′,7′-dichlor-fluorescein (DCF), carnitine, trypan blue, rhodamine123, N-(2-hydroxyethyl) piperazine-N′-(2-ethanesulfonic acid) (HEPES), bovine serum albumin (BSA), Hank’s Balanced Salt Solution (HBSS), dimethyl sulfoxide (DMSO), sucrose, D-mannitol, 2-amino-2-hydroxymethyl-propane-1,3-diol (TRIS), monopotassium phosphate, rotenone, 4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 3-morpholinopropane-1-sulfonic acid (MOPS), sodium succinate, magnesium chloride, potassium chloride, and GA were purchased from Sigma (St. Louis, MO USA). Celecoxib with CAS number 169590-42-5 and a purity of about 99% was gifted from Zahravi Pharmaceutical Co. (Tabriz, Iran). Gallic acid and celecoxib were freshly prepared before use and dissolved in ethanol.
Animal maintenance
Five male Wistar rats (body weight 200–220 g), originally from the animal house of the Baqiyatallah University of Medical Sciences (Tehran, Iran), were used in these experiments. The rats were kept under a daily 12 h light/dark cycle with controlled temperature and humidity (average of 22 C°, 50–60% relative humidity). Rats were allowed free access to water and food. Rats were adapted for 2 weeks before the isolation of cardiomyocytes and mitochondria. All experimental assays were carried out in conformity with the Guide for the Care and Use of Laboratory Animals approved by the Ethics Committee of the Ardabil University of Medical Sciences (Ardabil, Iran) with ethics code IR.ARUMS.REC.1398.284.
Solutions and buffers
Powell medium contained: 0.19 g KCl (2.5 mM), 6.43 g NaCl (110 mM), 0.3 g MgSO4 7H2O (1.2 mM), 5.96 g Hepes (25 mM), 0.16 g KH2PO4 (1.2 mM), and 1.98 g D (+)-glucose monohydrate (10 mM) in aqua sterile, pH adjusted to 7.4 with NaOH (2 mM) in a sterile medium. Calcium chloride (CaCl2) contained 100 mM CaCl2. Creatine-carnitine-taurine medium (CCT medium) contained 395.4 mg carnitine (2 mM), 655.5 mg creatine (5 mM), 625.5 mg taurine (5 mM), 3.6 g Hepes, and 10 µM cytosine β-D-arabinofuranoside and pH adjusted to 7.4 with NaOH (2 mM) in a sterile medium. Mitochondrial isolation buffer contained 75 mM sucrose, 0.2 mM EDTA, and 225 mM D-mannitol, pH adjusted to 7.4 with NaOH (2 mM). Mitochondrial assay buffer contained 140 mmol/L KCL, 0.5 mmol/L KH2PO4, 10 mmol/L NaCl, 0.5 mmol/L EGTA, 2 mmol/L MgCl2, 20 mmol/L HEPES, supplemented with 10 mmol/L succinate and 1 mg/mL rotenone, pH adjusted to 7.4. Mitochondrial swelling buffer contained 2 mmol/L MgCl2, 140 mmol/L KCL, 0.5 mmol/L KH2PO4, 10 mmol/L NaCl, 0.5 mmol/L EGTA, and 20 mmol/L HEPES, supplemented with 1 mg/mL rotenone, pH adjusted to 7.4.
Isolation of rat heart cardiomyocyte
Cardiomyocytes were isolated from male Wistar rats as previously described by Nippert et al. 20 After deep anesthesia with a combination of ketamine (50 mg/kg) and xylazine (10 mg/kg), hearts were quickly excised and directly perfused on a Langendorff perfusion system with Powell medium for 5 min and then replaced with enzymatic solution for 25 min. The hearts were removed from the apparatus and were dissected mechanically to small pieces and shaken in the enzymatic solution for 10 min. The digested cell suspensions were filtered through a mesh (180 µm) to remove additional tissues. The obtained cell suspensions were centrifuged at 1000 x g for 10 min. Isolated cardiomyocytes were suspended in CCT medium supplemented with 10% FBS and antibiotics (100 µg/ml penicillin and streptomycin) at 37°C under a 5% CO2−95% air atmosphere. The culture medium was changed to remove unattached dead cells, 1 h after plating.
Cell viability assay
The MTT assay provides sensitive measurements of the normal metabolic status of cells, where measurements reflect cell survival and mitochondrial function. 21 Rat heart–isolated cardiomyocytes (1 × 104 cells/well in 96-well plates) were incubated at 37°C for 6 h with different concentrations of celecoxib (0, 10, 25, 50, and 100 μg/mL). A negative control containing only cells and a solvent control (cardiomyocytes treated with 0.5% ethanol) was also evaluated. Also, for evaluation of ameliorative effect of GA, rat heart–isolated cardiomyocytes were treated with ethanol (0.05%) as control group, IC50 6h celecoxib, IC50 6h celecoxib + 10 µM GA, IC50 6h celecoxib + 50 µM GA, IC50 6h celecoxib + 100 µM GA and 100 µM GA at 37°C for 6 h. After treatment, the wells were incubated in the MTT solution (25 µL with final concentration of 0.5 mg/mL) for 2 h. The formed formazan crystals in intact cardiomyocytes were dissolved with DMSO (100 µL/well), and the absorbance at 570 nm was measured with an ELISA plate reader. All assays were performed in triplicate and the data were presented as the percentage of MTT reduction relative to the absorbance measured from negative control cells.
Isolation of rat heart mitochondria
Rat heart mitochondria were isolated from the male Wistar rats by the standard technique. 8 After deep anesthesia with a combination of ketamine (50 mg/kg) and xylazine (10 mg/kg), hearts were quickly excised, chopped, cleared from blood vessels, and homogenized with a glass homogenizer in a 10-fold volume of the mitochondrial isolation buffer at 4°C. The homogenized heart tissue was centrifuged at 1000× g for 10 min, and the pellet was removed. The obtained supernatant containing mitochondria was sedimented at 10,000× g for 10 min at 4°C. The Bradford assay was used for measurement of the protein content in mitochondria. Protein concentration in the mitochondrial suspension was 1000 µg/mL. Isolated mitochondria were kept for 2 h at 4°C.
Succinate dehydrogenase activity
Succinate dehydrogenase (SDH) activity was measured by the MTT assay where formazan purple color formation reflects mitochondrial function. 21 Rat heart–isolated mitochondria (100 µg/well in 96-well plates) were incubated at 37°C for 60 min with different concentrations of celecoxib (0, 1, 10, 25, 50, and 100 μg/mL). A negative control containing only mitochondria and ethanol (0.5%) as solvent of celecoxib and GA was also evaluated. Also, for evaluation of ameliorative effect of GA, rat heart–isolated mitochondrial were treated with ethanol (0.5%) as control group, IC50 1h celecoxib, IC50 1h celecoxib + 10 µM GA, IC50 1h celecoxib + 50 µM GA, IC50 1h celecoxib + 100 µM GA and 100 µM GA at 37°C for 60 min. After treatment, the mitochondria were incubated in the MTT solution (25 µL with final concentration of 0.5 mg/mL) for 30 min. The formed formazan crystals were dissolved with DMSO (100 µL/well), and the absorbance at 570 nm was measured with an enzyme-linked immunosorbent assay (ELISA) plate reader. All assays were performed in triplicate, and the data were presented as the percentage of MTT reduction relative to the absorbance measured from negative control.
Mitochondrial swelling assay
Mitochondrial swelling was estimated by examining changes in light scattering as monitored spectrophotometrically at 540 nm, where decrease in the absorbance reflect swelling of the mitochondrial and the mitochondrial permeability transition pore (PTP) opening. 22 Briefly, rat heart–isolated mitochondria (100 µg/well in 96-well plates) were treated with ethanol (0.05%) as control group, IC50 1h celecoxib, IC50 1h celecoxib + 10 µM GA, IC50 1h celecoxib + 50 µM GA, IC50 1h celecoxib + 100 µM GA and 100 µM GA at 37°C for 60 min. During 60 min, mitochondrial swelling was monitored by measurement of light scattering at 540 nm with a BioTek microplate reader (US).
Measurement of mitochondrial membrane lipid peroxidation
Mitochondrial membrane lipid peroxidation was determined by the reaction of thiobarbituric acid (TBA) with malondialdehyde (MDA), the end product of lipid peroxidation. Briefly, rat heart–isolated mitochondria (1 mg/mL) were treated with ethanol (0.05%) as control group, IC50 6h celecoxib, IC50 6h celecoxib + 10 µM GA, IC50 6h celecoxib + 50 µM GA, IC50 6h celecoxib + 100 µM GA and 100 µM GA at 37°C for 6 h. After treatment, mitochondria were mechanically lysed by homogenizer in a tube containing 1 mL 0.1% (w/v) trichloroacetic acid (TCA). Mitochondria homogenate samples were mixed with TCA (20%) and 0.5% TBA and incubated in boiling water bath for 15 min. After cooling on ice, the absorbance was measured at 532 nm. 23
Mitochondrial ROS level assay
The mitochondrial ROS formation was measured through flow cytometry by using DCF. Rat heart–isolated mitochondria (0.5 mg protein per mL) were incubated with ethanol (0.05%) as control group, IC50 1h celecoxib, IC50 1h celecoxib + 10 µM GA, IC50 1h celecoxib + 50 µM GA, IC50 1h celecoxib + 100 µM GA and 100 µM GA in respiration buffer containing 10 mM Tris, 0.32 mM sucrose, 50 mM EGTA, 20 mM Mops, 0.5 mM MgCl2, 5 mM sodium succinate and 0.1 mM KH2PO4 pH 7.4) for 60 min at 37°C. The samples with DCF (10 μM) were incubated for 15 min and then determined through flow cytometry (Cyflow Space-Partec, Germany). The signals were obtained using a 530 nm bandpass filter (FL-1 channel). Each determination is based on the mean fluorescence intensity of 20,000 counts. The data were analyzed by FlowJo software. 24
Mitochondrial membrane potential collapse assay
The mitochondrial membrane potential (MMP) collapse was measured through flow cytometry by using rhodamine 123. Rat heart–isolated mitochondria (0.5 mg protein per mL) were incubated with ethanol (0.05%) as control group, IC50 1h celecoxib, IC50 1h celecoxib + 10 µM GA, IC50 1h celecoxib + 50 µM GA, IC50 1h celecoxib + 100 µM GA and 100 µM GA in MMP buffer containing 220 mM sucrose, 5 mM KH2PO4, 68 mM D-mannitol, 50 μM EGTA, 10 mM HEPES, 5 mM sodium succinate, 2 mM MgCl2, 10 mM KCl, and 2 μM rotenone for 60 min at 37°C. The samples with rhodamine 123 (5 μM) were incubated for 15 min and then determined through flow cytometry (Cyflow Space-Partec, Germany). The signals were read on FL-1 channel. Each determination is based on the mean fluorescence intensity of 20,000 counts. The data were analyzed by FlowJo software. 8
Statistical analysis
Results were expressed as mean ± standard deviation (SD). Result analyses were performed using Graph Pad Prism 5.0 software (Graph Pad, San Diego, CA). Results were analyzed by one- and two-way analysis of variance (ANOVA); Tukey’s and Bonferonie’s tests were applied for post hoc analysis. A value of p < .05 was considered to be statistically significant.
Results
Effect of GA on celecoxib-induced cytotoxicity in isolated cardiomyocytes
Celecoxib treatment (25, 50 and 100 μg/mL) significantly (p < .001) decreased the cell viability when compared to untreated control (Figure 1(a)). Co-treatment of GA (50 and 100 µM) with celecoxib (16 µg/ml) significantly (p < .001) increased the cell viability compared to celecoxib-treated group, whereas GA (100 µM) treatment did not show significant changes in the cell viability compared to control group (Figure 1(b)). Cell viability of rat heart isolated–cardiomyocytes after treatment for 6 h with different concentrations celecoxib (A) and celecoxib, celecoxib + GA with different concentrations and GA alone (B). Rat heart–isolated cardiomyocytes viability was determined using the MTT assay. Values are expressed as the percentage of viable cells, with the viability of untreated control cells set at 100%. Values represent mean ± SD (n = 3) of three independent experiments, one-way ANOVA, Tukey’s test. ***p < .001, relative to untreated cells. ###p < .001, relative to celecoxib treated cells. GA, gallic acid; MTT, 2,5-diphenyltetrazolium bromide.
Effect of GA on celecoxib-induced mitochondrial dysfunction in isolated mitochondria
Succinate dehydrogenase activity as marker of mitochondrial function was significantly (p < .001) decreased in celecoxib-treated group when compared to untreated control in isolated mitochondria (Figure 2(a)). Co-treatment of GA (10, 50 and 100 µM) with celecoxib (16 µg/ml) significantly (p < .001) increased SDH activity compared to celecoxib-treated group, whereas GA (100 µM) treatment did not show significant effect on SDH activity compared to control group (Figure 2(b)). Succinate dehydrogenase activity of rat heart–isolated mitochondria after treatment for 60 min with different concentrations celecoxib (A) and celecoxib, celecoxib + GA with different concentrations and GA alone (B). Mitochondrial succinate dehydrogenase activity was determined using the MTT assay. Values are expressed as the percentage of intact and active mitochondria, with the activity of untreated control mitochondria set at 100%. Data are mean ± SD (n = 3) of three independent experiments, one-way ANOVA, Tukey’s test. ***p < .001, relative to untreated mitochondria. ###p < .001, relative to celecoxib-treated mitochondria. SDH, succinate dehydrogenase; GA, gallic acid; MTT, 2,5-diphenyltetrazolium bromide.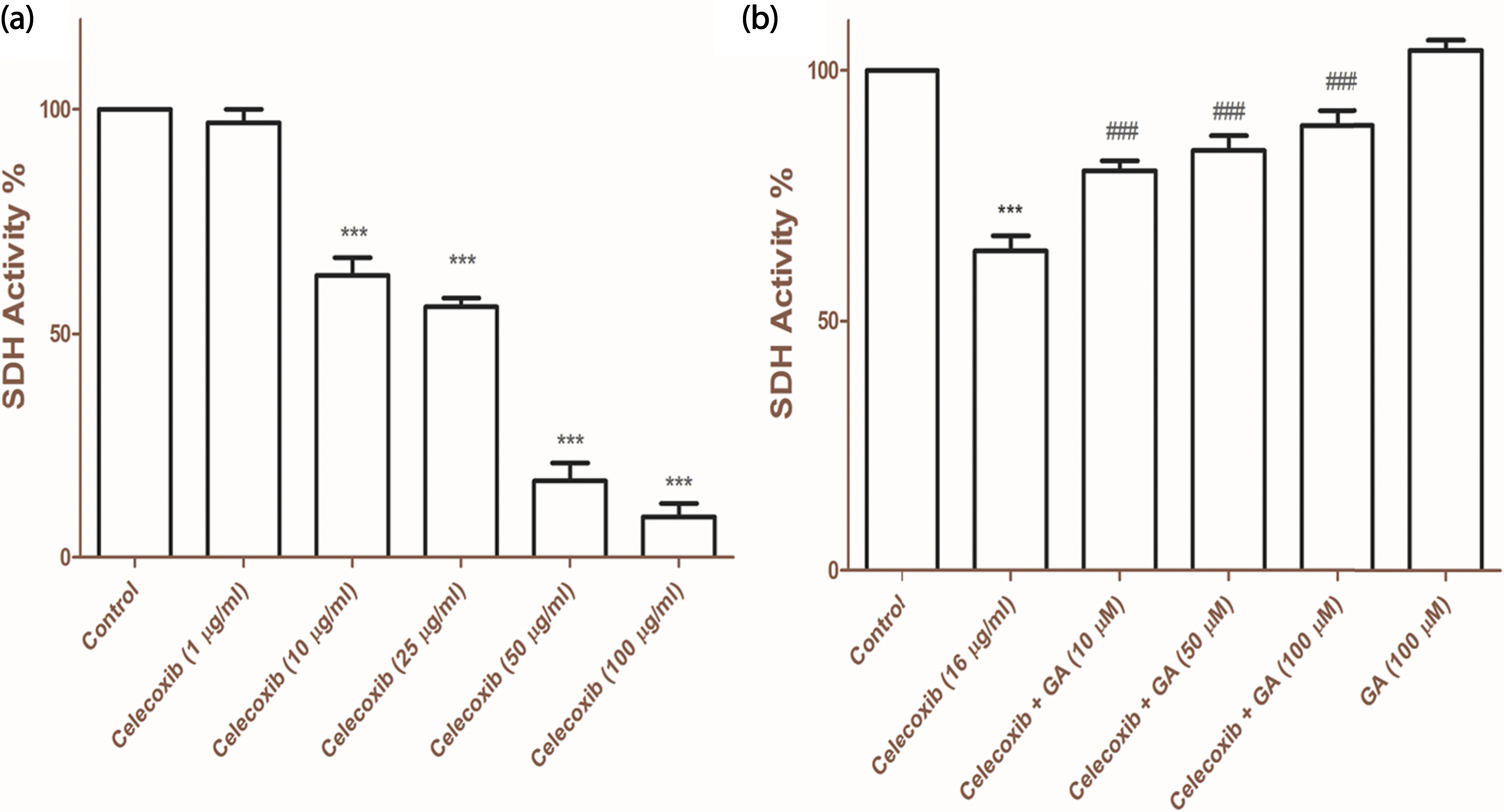
Effect of GA on celecoxib-induced mitochondrial lipid peroxidation in isolated mitochondria
Malondialdehyde levels were significantly (p < .001) increased in celecoxib-treated group when compared to untreated control. Co-treatment of GA (10, 50 and 100 µM) with celecoxib (16 µg/ml) significantly (p < .001) restored the levels of MDA compared to celecoxib-treated group, whereas GA (100 µM) treatment did not show significant changes in the MDA levels compared to control group (Figure 3(a)). Mitochondrial lipid peroxidation (A) and mitochondrial swelling (B) of rat heart–isolated mitochondria after treatment for 60 min with celecoxib, celecoxib + GA with different concentrations and GA alone. Mitochondrial lipid peroxidation was determined by the reaction of TBA with MDA. Mitochondrial swelling was determined by monitoring the absorbance of mitochondrial suspension at 540 nm. The decrease in absorbance reflects mitochondrial swelling and MPT pore opening. Values are expressed as mean ± SD (n = 3) of three independent experiments, one-way ANOVA, Tukey’s test (A) and two-way ANOVA followed, Bonferroni test (B). ***p < .001, relative to untreated mitochondria. ###p < .001, relative to celecoxib-treated mitochondria. TBA, thiobarbituric acid; MPT, Mitochondrial Permeability Transition; MDA, malondialdehyde; GA, gallic acid.
Effect of GA on celecoxib-induced mitochondrial swelling in isolated mitochondria
Mitochondrial swelling was significantly (p < .001) increased in celecoxib-treated group when compared to untreated control in isolated mitochondria. Our data indicated that in the presence of celecoxib, absorbance at 540 nm markedly decreased. Co-treatment of GA (100 µM) with celecoxib (16 µg/ml) significantly (p < .001) restored mitochondrial swelling and increased the absorbance at 540 nm compared to celecoxib-treated group, whereas GA (100 µM) treatment did not show significant changes in the MDA levels compared to control group. However, GA (10 and 50 µM) treatment failed to increase the absorbance at 540 nm and decrease mitochondrial swelling in isolated mitochondria (Figure 3(b)).
Effect of GA on celecoxib-induced mitochondrial ROS formation in isolated mitochondria
The rates of ROS formation in heart mitochondria isolated from rats treated with celecoxib, celecoxib + GA, and GA are shown in Figure 4. The rate of ROS formation in the mitochondrial suspension was determined through the changes in the fluorescence intensity of DCF. Mitochondrial ROS formation was significantly (p < .001) increased in celecoxib-treated group when compared to untreated control in isolated mitochondria. Co-treatment of GA (10, 50, and 100 µM) with celecoxib (16 µg/ml) significantly (p < .001) decreased ROS formation compared to celecoxib-treated group. Our study showed that in presence of GA, celecoxib-induced ROS formation was decreased in a concentration-dependent manner, whereas GA (100 µM) treatment did not show significant effect on mitochondrial ROS formation compared to control group. Mitochondrial ROS formation of rat heart isolated mitochondria after treatment for 60 min with celecoxib, celecoxib + GA with different concentrations and GA alone. The rate of ROS formation in the mitochondrial suspension was determined through the changes in the fluorescence intensity of DCF. 2′,7′-Dichlor-fluorescein fluorescence was monitored by flow cytometer and fluorescent signals were displayed as histogram and fluorescence intensity mean (A). Summary of ROS formation (B). Values are expressed as mean ± SD (n = 3) of three independent experiments, one-way ANOVA, Tukey’s test. ***p < .001, relative to untreated mitochondria. ###p < .001, relative to celecoxib-treated mitochondria. ROS, reactive oxygen species; DCF, 2′,7′-dichlor-fluorescein; GA, gallic acid.
Effect of GA on celecoxib-induced mitochondrial membrane potential collapse in isolated mitochondria
The rates of MMP collapse in heart mitochondria isolated from rats treated with celecoxib, celecoxib + GA, and GA are shown in Figure 5. The rate of MMP collapse in the mitochondrial suspension was determined through the changes in the fluorescence intensity of rhodamine 123. Mitochondrial membrane potential collapse was significantly (p < .001) increased in celecoxib-treated group when compared to untreated control in isolated mitochondria. Co-treatment of GA (10, 50, and 100 µM) with celecoxib (16 µg/ml) significantly (p < .001) decreased MMP collapse compared to celecoxib-treated group. Our study showed that in presence of GA, celecoxib-induced MMP collapse was decreased in a concentration-dependent manner, whereas GA (100 µM) treatment did not show significant effect on collapse compared to control group. Mitochondrial membrane potential collapse of rat heart isolated mitochondria after treatment for 60 min with celecoxib, celecoxib + GA with different concentrations and GA alone. The rate of MMP collapse in the mitochondrial suspension was determined through the changes in the fluorescence intensity of rhodamine 123. Rhodamine 123 fluorescence was monitored by flow cytometer and fluorescent signals were displayed as histogram and fluorescence intensity mean (A). Summary of MMP collapse (B). Values are expressed as mean ± SD (n = 3) of three independent experiments, one-way ANOVA, Tukey’s test. ***p < .001, relative to untreated mitochondria. ###p < .001, relative to celecoxib-treated mitochondria. MMP, mitochondrial membrane potential; GA, gallic acid.
Discussion
However, the Food and Drug Administration (FDA) has approved celecoxib to remain for clinical applications on the market after concluding that the potential risk of celecoxib is lower than its benefit. It has been reported that the cardiovascular risk profile of celecoxib is much low and even minor. 25 Nevertheless, celecoxib may cause a significant increase in serious cardiovascular events, such as congestive heart failure, stroke, and myocardial infarction. 3 Serious cardiovascular events were reported in 2.5% of patients receiving celecoxib compared to 1.9% in the placebo group. 26 It seems that the risk of cardiovascular events induced by celecoxib is associated to the dose and the dosing interval. It has been reported that celecoxib 400 mg twice daily displayed a greater than three-fold risk for combined endpoints of stroke, myocardial infarction, heart failure, and cardiovascular death compared with placebo and 200 mg twice daily with a greater than two-fold risk. 27 Circulating concentrations of celecoxib have been detected in pharmacokinetic human. Chow et al. have reported that administration of 400 mg celecoxib daily to 68 healthy adults for 2 weeks led to a mean plasma concentration of 607 ± 338 ng/mL (range 82–1700 ng/mL). 28 Celecoxib is rapidly absorbed and achieves peak serum concentration in approximately 3 h. Therefore, the possibility of exposure of different cells such as cardiomyocytes and intracellular organs such as mitochondria to celecoxib will be high because past studies have demonstrated accumulation of celecoxib in several cell lines due to its integration into the hydrophobic core of cellular phospholipid membranes. 29 Due to human exposure and serious cardiovascular events of celecoxib, more studies to understand the mechanism of its cardiotoxicity and suggest a preventative solution to reduce its toxicity can be helpful. Therefore, the present study explored the cellular and mitochondrial toxicity of celecoxib along and in presence GA as an antioxidant in the cardiomyocytes and cardiac mitochondria. Our results on rat isolated–cardiomyocytes showed that celecoxib causes cytotoxicity and decreases the cell viability. Sakane et al. have proved that celecoxib reduces cell viability in a dose-dependent manner in H9c2 cardiac cell line and obtained data of their study is consistent with our result in the current study. 9 Furthermore, the effect of celecoxib on isolated mitochondria was accompanied with mitochondrial dysfunction, ROS formation, MMP collapse, mitochondrial swelling, and lipid peroxidation. Our results showed that celecoxib can induce toxicity in cardiac mitochondria. Previous studies have reported that celecoxib and other NSAIDs can cause mitochondrial dysfunction and oxidative stress.3,7 The results obtained in previous studies are in line with our data in this study.3,7 The main mechanism by which these drugs can cause mitochondrial dysfunction and oxidative stress in cardiomyocytes is not fully understood, but preventing increased mitochondrial ROS levels and mitochondrial dysfunction by antioxidant and mitochondrial protective agents may reduce the toxic effects of increased ROS levels and reduce the incidences of cardiotoxicity caused by celecoxib and NSAIDs. 3
Today, there are different experimental studies about identification of the intracellular targets and the molecular mechanisms involved in NSAID-induced cardiovascular events.3,30 These studies indicated that a multifactorial process is involved in NSAID-induced cardiotoxicity.3,30 However, in fact, mitochondrial dysfunction has become an important hallmark of NSAID-induced cardiovascular events. 31 Previous studies and our results in the current work have shown abnormalities in mitochondrial functions such as defects in the SDH activity (mitochondrial complex II), ROS formation, mitochondrial swelling, MMP collapse, lipid peroxidation, and a vicious cycle of free radical formation.8,31 Due to the high energy demand of the heart, in comparison with other tissues, mitochondria are abundant in heart tissue and about constituting 45% of the cardiomyocytes volume. 32 Accordingly, it can be anticipated that the development of antioxidant and mitochondrial protective agents is a rational strategy to prevent drug-induced oxidative damage and cardiotoxicity. 33 Therefore, mitochondrial protection from oxidative damages caused by drugs and chemicals can play an effective role in inhibiting cardiomyocytes death and ultimately cardiotoxicity. 34 Gallic acid as a plant phenolic compound shows a good antioxidant potential via chelating pro-oxidant transition metals, scavenging free radicals by electron transfer and/or hydrogen donation, inhibiting lipid peroxidation processes as well as several pro-oxidant enzymes involved in reactive oxygen species (ROS) formation.12,35 It has been reported that mitochondrially targeted GA can be used for preventing mitochondrial impairment caused by oxidative stress after exposure with sodium nitroprusside in brain isolated mitochondria. 36 Dutta and Paul showed that GA protects liver mitochondria probably by reducing the oxidative stress against bisphenol A. 19 Besides, they reported that GA can protect the mitochondrial surface from bisphenol A–induced oxidative damages. 19 Our results also showed that GA can protect mitochondrial dysfunction, ROS formation, lipid peroxidation, mitochondrial swelling induced by celecoxib in isolated mitochondria, and these results were confirmed in cellular evaluations. Recent study has shown that a new mitochondriotropic antioxidant (AntiOxBEN3) based on the dietary antioxidant GA can inhibit MPT pore opening, 33 which can play an effective role in reducing mitochondrial dysfunction, swelling and MMP collapse, which is consistent with obtained results in the current study. We showed that GA inhibits celecoxib-induced MPT pore opening, mitochondrial swelling, and ROS formation and reduces celecoxib toxicity in isolated cardiomyocytes and mitochondria.
In conclusion, the data of this study showed that celecoxib can directly cause toxicity in cardiac mitochondria and cardiomyocytes, which is associated with cytotoxicity, ROS formation, oxidative stress, mitochondrial dysfunction, which is significantly attenuated by GA. Gallic acid inhibits celecoxib-induced MPT pore opening, mitochondrial swelling, and ROS formation and reduces celecoxib toxicity in isolated cardiomyocytes and mitochondria. Due to GA availability as a supplement, the absence of toxic side effects, and good effects on cardiovascular system, this phenolic compound deserves to be a promising candidate in clinical trial studies to reduce celecoxib cardiotoxicity. Due to hydrophilic nature of GA, its bioavailability and distribution is low throughout the body with the inherent difficulties to cross cellular membranes and mitochondria. Considering that the results of the current study are direct effect of GA on isolated cardiomyocytes and mitochondria, therefore, it is suggested its low bioavailability and distribution be considered in future studies.
Footnotes
Declaration of Conflicting Interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Ardabil University of Medical Sciences, Deputy of Research with ethics code IR.ARUMS.REC.1398.284.