
Abstract
Allergic rhinitis (AR) is a type I hypersensitive disease. Long non-coding RNA (lncRNA) SNHG16 acts as an oncogene in a variety of tumors and promotes the occurrence of inflammation in many inflammatory diseases. The study aims to investigate the expression of SNHG16 and its potential biological functions in AR. RT-qPCR results showed that the expression of SNHG16 in AR was up-regulated. The AR cell model was constructed by stimulating primary nasal mucosal epithelial cells from AR patients with IL-13. After knocking down the expression of lncRNA SNHG16, cell apoptosis was detected by flow cytometry, and the expression of inflammatory factors was detected by ELISA. The results showed that SNHG16 promoted cell apoptosis and inflammation. Then, bioinformatics analysis was used to screen miRNAs bound with SNHG16. Luciferase reporter gene assay and RNA pull-down experiment were used to verify the relationship. We found that the expression of miR-106b-5p was down-regulated and leukemia inhibitory factor (LIF) expression was up-regulated in the AR cell model. The expression of phospho-Janus kinase 1 and p-signal transducer and activator of transcription 3 (STAT3) were detected by Western blotting. Silencing the expression of LIF could inhibit the activity of JAK1/STAT3 pathway and further inhibit cell apoptosis and the occurrence of inflammation. Then transfected SNHG16 shRNA alone or together with miR-106b-5p antagomir into the AR cell model, we found that silencing the expression of SNHG16 down-regulated the expression of LIF and inhibited the activity of the JAK1/STAT3 pathway, cell apoptosis, and inflammation. However, miR-106b-5p antagomir weakened its inhibitory effects. The role of SNHG16 in AR was further verified by the ovalbumin-induced AR mouse model in vivo. In conclusion, SNHG16 up-regulates LIF expression by binding with miR-106b-5p, thus promoting the activity of JAK1/STAT3 pathway, and promoting the development of AR. These results provide new targets for the treatment of AR and may help reduce the damage caused by AR.
Keywords
Introduction
The long-term inflammation of nasal mucosa is regulated by non-infectious immunoglobulin E (IgE) and further regulated by interleukin-13 (IL-13).1,2 IL-13 is a typical T helper (Th)2 cytokine, which is the central mediator of IgE-mediated allergic diseases. The clinical manifestations of allergic rhinitis (AR) are runny nose, sneeze, itchy nose, and nasal congestion. 3 This disease not only reduces people’s quality of life and daily work, but also has a negative impact on social economy. 4 In recent years, a growing body of evidence indicates the functions of non-coding RNA (ncRNA) in various physiological and pathological processes, but there has been little research on AR.
The long non-coding RNA (lncRNA) SNHG16 is a small nucleolar RNA host gene 16. It is the first lncRNA found in neuroblastoma. 5 It has been shown that lncRNA SNHG16 was highly expressed in many cancers, such as colorectal adenomas/adenocarcinomas, 6 hemangioma, 7 bladder cancer, 8 and breast cancer. 9 It is reported that SNHG16 inhibited cell viability and promoted apoptosis and inflammatory injury by targeting the miR-370-3p/IGF2 axis in lipopolysaccharide (LPS)-induced acute pneumonia in A549 cells. 10 Therefore, we investigated the effect and mechanism of SNHG16 on the progression of AR.
Few studies have found that miRNAs may be involved in the pathogenesis of AR.11,12 Leukemia inhibitory factor (LIF) is a pleiotropic glycoprotein that belongs to IL-6 family. It has a wide range of biological activities, including promoting the growth and differentiation of different type of cells13,14 and affecting inflammation. 15 LIF has a variety of functional activities and is involved in the pathogenesis and development of various diseases. 16 Some studies have reported that LIF is a pro-inflammatory mediator of various inflammatory diseases, 17 and some studies have pointed out that LIF has anti-inflammatory effects. 18 Our research mainly studied the effect and mechanism of LIF on inflammation in AR.
Th lymphocytes play an important role in the sensitization process. According to the secreted cytokines, they divided into two categories, Thl and Th2. Thl cells antagonize the IgE response and inhibit IgE production by secreting IL-2 and interferon γ (INF-γ), while Th2 cells produce IL-4, IL-5, and other cytokines that promote IgE production. They antagonize each other and maintain the balance of Thl and Th2. The imbalance of Thl and Th2 cytokine network is an important cause of AR. IL-4, also known as B cell growth factor, can promote Th0 to Th2 differentiation, increase the expression of endothelial cell adhesion molecules, inhibit eosinophil apoptosis, and promote the growth and differentiation of B cell and secretion of IgE antibodies. 19 Cell adhesion molecule is a type of glycoprotein that mediates the interaction between cell and cell and cell and extracellular matrix. And it plays an important role in many physiological and pathological processes, such as embryo differentiation, normal tissue structure, inflammatory, and immune response. 20 Vascular cell adhesion molecule-1 (VCAM-1), which leads to the migration of eosinophils through specific binding with ligands on the surface of eosinophils, is an important part of the pathogenesis of allergy. We judged the progress of AR by detecting the secretion of the above factors.
Here, the AR cell model was constructed by stimulating primary nasal mucosal epithelial cells (NECs) from AR patients with IL-13. The aim of this study was to investigate the function and molecular mechanism of lncRNA SNHG16 in AR. These results may provide new targets for the treatment of AR.
Materials and methods
Collection of AR tissue samples
The AR nasal mucosal tissue samples were collected from 20 patients (16∼48 years old) with AR, and the normal tissue samples were obtained from 20 patients with nonallergic rhinitis (NAR, 23∼57 years old) in our hospital. The symptom scores for patients with allergic rhinitis were shown in Supplementary Table II. Partial inferior turbinectomy was conducted to relieve the nasal obstruction in the AR and NAR patients with nasal septum deviation, and samples were obtained from the inferior turbinate sections. And there were no complications during the operation. All procedures performed in this study were performed in compliance with ethical guidelines approved by the Institutional Review Board and Human Ethics Committee of the First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, China (approval number: 2019-1847), and in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants.
Cell culture and treatment
Primary NECs were isolated from the inferior turbinate tissues of allergic rhinitis patients and maintained in Bronchial Epithelial Growth Medium (BEGM, Lonza, Walkersville, MD, USA).11,21 Human nasal mucosal epithelial cells (HNEpCs) and human embryonic kidney 293T (HEK-293T) cells were maintained in DMEM containing 10% fetal bovine serum and cultured in a 37°C incubator containing 5% CO2. Then the primary NECs were stimulated with IL-13 (50 ng/mL) to construct the AR cell model.11,21 After 24 h of culture, we transfected SNHG16 interference vector (the final concentration was 5 μg/mL), miR-106b-5p antagomir (the final concentration was 30 nM), or LIF siRNA (the final concentration was 50 nM) into the cells by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
RT-qPCR
Trizol reagent (Takara, Dalian, China) was used to extract RNA from tissue and cell samples. After measuring the concentration, cDNA was synthesized by the same amount of RNA by using Takara RT reagent kit. Then RT-qPCR reaction was performed by using the cDNA as a template. The procedure was as follows: 95°C for 30 s followed by 40 cycles of 95°C for 5 s and 60°C for 30 s. miR-106b-5p used U6 as an internal reference, and mRNA used β-actin as an internal reference. The primers used in the experiment were designed and synthesized by Invitrogen. The obtained data were calculated by the 2−ΔΔCt method.
Cell apoptosis assay
Cell apoptosis was detected by using Annexin V-FITC/PI kit (Vazyme, Nanjing, China). After 48 h of transfection, cells were digested with trypsin and suspended by using Annexin V binding buffer to a final concentration of 1 × 106 cells/Ml and then added 5 μL of Annexin V-FITC and 5 μL of PI to 100 μL of cell suspension and incubated at room temperature for 15 min. Finally, 400 μL of Annexin V binding buffer was added for flow cytometry detection.
Luciferase reporter gene assay
Bioinformatics analysis was used to predict the miRNA bound with SNHG16 and its downstream target gene. The binding sites of wild-type (WT) and mutant (MUT) SNHG16 or the 3’-UTR binding sites of WT or MUT LIF luciferase reporter vectors were constructed and then co-transfected with miR-106b-5p-mimic into HEK-293T cells. Luciferase activity was determined with a Dual-Luciferase® Reporter Assay System (Promega) 48 h later.
RNA pull-down assay
In order to further verify the relationship between lncRNA SNHG16 and miR-106b-5p, we synthesized biotin-labeled miR-106b-5p (Bio-miR-106b-5p) for RNA pull-down assay. Cells were transfected with Bio-miR-106b-5p or Bio-NC-mimic (8 nM) for 48 h, then washed with PBS, and lysed with the lysis buffer. The lysate was incubated with avidin-anchored magnetic beads at 4°C for 3 h. Then the beads were washed by the lysis buffer, washed three times with low salt buffer and once with high salt buffer. The RNA complex was eluted, and the RNA pull-down product was subjected to specific RT-qPCR detection.
Western blot analysis
Cell samples were lysed on ice with RIPA lysis buffer (Solarbio, Beijing, China), and the supernatant was collected by centrifugation. After determining the concentration by BCA kit (Beyotime Institute of Biotechnology, Shanghai, China), the equal amount of protein sample was taken for SDS-PAGE in 70 V for 30 min and 120 V for 90 min and then transferred the corresponding size protein bands to polyvinylidene difluoride membranes at 300 mA for 2 h. After blocking in skimmed milk for 2 h at room temperature, the membranes were then incubated with the following antibodies at 4°C overnight. The antibody against cleaved caspase 3 (1:500, ab2302), LIF (1:1000, ab113262), phospho-Janus kinase1 (p-JAK1) (1:1500, ab138005), JAK1 (1:2000, ab47435), p-signal transducer and activator of transcription 3 (STAT3) (1:1500, ab30647), STAT3 (1:2000, ab119352), and GAPDH (1:2000, ab9485) was purchased from Abcam. Anti-Bcl-2 antibody (1:1000, #15071) was purchased from Cell Signaling Technology. After washing with TBS-T, the membranes were incubated with the horseradish peroxidase-conjugated IgG antibody, and enhanced chemiluminescence kit (Millipore, MA, USA) was used to detect the protein bands.
Enzyme-linked immunosorbent assay (ELISA)
After 48 h of transfection, the cell culture supernatant of each group was obtained by centrifugation. The secretion of IgE, IL-4, IL-10, and sVCAM-1 was detected by human IgE ELISA Kit (ab108650), human IL-4 ELISA Kit (ab215089), human IL-10 ELISA Kit (ab100549), and human VCAM-1 ELISA Set (ab47355) according to the manufacturer’s instructions. The absorbance was measured by a microplate reader (Thermo Fisher Scientific, Waltham, Massachusetts, USA) at 450 nm. The results were calculated according to the standard curve.
Immunofluorescence staining
After washing with PBS, the cell was fixed with 4% paraformaldehyde for 20 min and then permeabilized with 0.5% Triton X-100 for 10 min. Cells were incubated with 3% BSA for 1 h to block non-specific immunoglobulin binding and incubated with rabbit monoclonal to occludin antibody (1:50, ab216327, Abcam) and rabbit monoclonal to zonula occludens-1 (ZO-1) antibody (1:50, ab221547, Abcam) at 4°C overnight. The secondary antibodies were goat anti-rabbit IgG H&L (Alexa Fluor 488) (1:200, ab150077, Abcam) and goat anti-rabbit IgG H&L (Alexa Fluor 546) (1:200, A-11035, Invitrogen). The nuclei were stained with 4’-6-diamidino-2-phenylindole dihydrochloride for 5 min. The images were then observed and collected under a fluorescence microscope.
In vivo experiment
To further clarify the role of SNHG16 in AR, the mice model of allergic rhinitis was established by intraperitoneally injecting with nasal drip. First, thirty 8-week-old female BALB/c mice in the AR group were intraperitoneally injected with 100 μL of sensitization fluid containing 0.5 mg/mL ovalbumin (OVA) and 20 mg/mL Al(OH)3 on day 1, 8, and 15. And 10 mice in the control group were injected intraperitoneally with 100 μL of 0.9% normal saline as a negative control. After 1 week, the AR model mice were divided into three groups: OVA group, OVA+sh-NC group, and OVA+sh-SNHG16 group. Negative control shRNA or lncRNA SNHG16 shRNA was intranasally instilled 2 h before nasal provocation. Then the mice were stimulated by nasal instillation of OVA solution (2 mg/mL); each mouse was instilled with 50 μL once a day for 1 week. The mice in the control group were also used normal saline as a negative control. Then the allergic symptoms in mice in different groups were scored. After successful modeling, the mice were sacrificed on the second day of the last nasal challenge, and then the blood and nasal mucosa specimens were collected for further experiments. The tissues were removed and immediately fixed in 4% paraformaldehyde for 24 h at 4°C, followed by dehydration in a series of increasing concentrations of ethanol and embedded in paraffin. The 4 μm-thick sections were deparaffinized and stained with hematoxylin and eosin (H&E) for histopathological examination. The secretion of IgE, IL-4, IL-10, and sVCAM-1 in serum was detected by ELISA kits according to the manufacturer’s instructions. All animal procedures were performed in accordance with the Principles of Laboratory Animal Care (NIH) and approved by the Ethical Committee on Animal Care and Use of the First Affiliated Hospital of Xi’an Jiaotong University (approval number: XJTULAC2019-2561).
Statistical analysis
Statistical analysis was performed by using SPSS software. Results were expressed as mean ± standard error of mean. Comparisons between two groups were performed by using the t-test. Comparisons between multiple groups were performed by using one-way analysis of variance (ANOVA). p < 0.05 indicated statistically significant difference.
Results
LncRNA SNHG16 was up-regulated in AR
To investigate the role of SNHG16 in AR, RT-qPCR was used to detect the expression level of SNHG16 in AR tissue samples. The expression of SNHG16 in AR tissue samples was significantly up-regulated than that in normal tissue samples (Figure 1(A)). We further stimulated the HNEpCs by IL-13 and stimulated the primary NECs from allergic rhinitis patients by IL-13 to obtain the AR cell model, then detected the expression of SNHG16 by RT-qPCR, and found that the expression of SNHG16 was up-regulated in the AR cell model (Figures 1(B) and (C)). SNHG16 was up-regulated in AR. (A) The expression of SNHG16 in tissue samples from 20 (16∼48 years old) AR patients and 20 (23∼57 years old) NAR patients was detected by RT-qPCR. n = 20. **p < 0.01, Student’s t-test. (B) The expression of SNHG16 in IL-13 stimulated HNEpCs was detected by RT-qPCR. n = 6. **p < 0.01, Student’s t-test. (C) After constructed the AR cell model by stimulating primary NECs isolated from the AR patients with IL-13, the expression of SNHG16 was detected by RT-qPCR. n = 6. **p < 0.01, Student’s t-test.
LncRNA SNHG16 promoted cell apoptosis and inflammation in AR
We next interfered the expression of SNHG16 in IL-13 stimulated HNEpCs (Figure 2(A)). Immunofluorescence staining results showed that the expression levels of tight junction proteins such as occludin and ZO-1 in HNEpCs treated with IL-13 were decreased, while knocking down SNHG16 could up-regulate the tight junction protein expression (Supplementary Figure 1). Then, the flow cytometry results showed that knockdown of SNHG16 inhibited cell apoptosis (Figure 2(B)), while the expression of anti-apoptotic protein Bcl-2 was up-regulated, and the cleaved caspase-3 expression was down-regulated (Figure 2(C)). As shown in Figures 2(D)–(G), after interfering the expression of SNHG16, the secretion of IgE, inflammatory factor IL-4, and cell adhesion factor sVCAM-1 was down-regulated, while the secretion of anti-inflammatory factor IL-10 was up-regulated. In order to understand the effects of SNHG16 on AR, we overexpressed or interfered the expression of SNHG16 in the AR cell model and then detected the cell apoptosis and inflammation responses. The results indicated that SNHG16 promoted cell apoptosis and inflammation of AR (Supplementary Figure 2). SNHG16 promoted cell apoptosis and inflammation of AR. Interfering the expression of SNHG16 in IL-13 stimulated HNEpCs, (A) RT-qPCR was used to detect the interference effect. (B) The effect of SNHG16 on cell apoptosis was measured by flow cytometry. (C) Western blotting was used to detect the expression of cleaved caspase-3 and anti-apoptotic proteins Bcl-2, and the gray value was quantitatively analyzed by Image J. (D)–(G) The secretion of IgE, inflammatory factor IL-4, anti-inflammatory factor IL-10, and cell adhesion factor sVCAM-1 in cell supernatant was detected by ELISA. n = 6. **p < 0.01, two-way ANOVA with Sidak’s correction.
LncRNA SNHG16 bound with miR-106b-5p
Then we predicted that SNHG16 bound with miR-106b-5p by using bioinformatics analysis (Figure 3(A)). We found that miR-106b-5p expression was down-regulated in IL-13 stimulated HNEpCs and the AR cell model (Figures 3(B) and (C)). Subsequently, miR-106b-5p mimic and antagomir were synthesized and transfected into the AR cell model to test the efficiency (Figure 3(D)). To verify the relationship between SNHG16 and miR-106b-5p, we performed the luciferase report gene assay in HEK-293T cells and found that miR-106b-5p mimic decreased the luciferase activity of WT SNHG16 group (Figure 3(E)). Then, in order to determine SNHG16 bound with miR-106b-5p directly, RNA pull-down assay was carried out with biotinylated miR-106b-5p (Bio-miR-106b-5p) probe to detect the expression of SNHG16. Compared with the negative control (Bio-NC-mimic), SNHG16 expression in the Bio-miR-106b-5p group was significantly up-regulated (Figure 3(F)). In addition, as shown in Figure 3(G), we overexpressed or interfered the expression of SNHG16 and then detected the expression of miR-106b-5p by RT-qPCR. We found that SNHG16 negatively regulated the expression of miR-106b-5p. Based on these results, we found that SNHG16 directly bound with miR-106b-5p, and their expression was negatively correlated. SNHG16 directly bound with miR-106b-5p and inhibited miR-106b-5p expression. (A) The sequence region of SNHG16 bound with miR-106b-5p. (B), (C) RT-qPCR was used to detect the expression of miR-106b-5p in IL-13 stimulated HNEpCs and AR cell model. n = 6. **p < 0.01, Student’s t-test. (D) After transfection with miR-106b-5p mimic or miR-106b-5p antagomir into AR cell model for 48 h, its efficiency was examined by RT-qPCR. n = 6. **p < 0.01, one-way ANOVA with Tukey’s correction. (E) Co-transfected WT or MUT SNHG16 luciferase reporter gene vector with miR-106b-5p-mimic into HEK-293T cells. After 48 h, the luciferase activity was measured by the luciferase reporter gene system. (F) The binding of SNHG16 with miR-106b-5p was verified by biotinylated miR-106b-5p RNA pull-down assay. (G) After overexpression or knockdown of SNHG16 in AR cell model, miR-106b-5p expression was detected by RT-qPCR. n = 6. *p < 0.05, **p < 0.01, one-way ANOVA with Tukey’s correction.
MiR-106b-5p directly targeted LIF
Next, in order to study the mechanism of miR-106b-5p in the AR cell model, LIF was predicted to be the target gene of miR-106b-5p by bioinformatics analysis (Figure 4(A)). LIF expression was up-regulated in IL-13 stimulated HNEpCs and the AR cell model (Figures 4(B) and (C)). Then, the targeting effect was verified by luciferase reporter gene assay in HEK-293T cells. The results showed that miR-106b-5p mimic down-regulated the luciferase activity of WT-LIF-3’-UTR (Figure 4(D)). In addition, in the AR cell model, miR-106b-5p mimic down-regulated LIF expression (Figure 4(E)). These results indicated that miR-106b-5p directly targeted LIF. MiR-106b-5p directly targeted LIF. (A) The binding site of miR-106b-5p targeted LIF 3’-UTR. (B), (C) The expression of LIF in IL-13 stimulated HNEpCs and AR cell model was detected by RT-qPCR. n = 6. **p < 0.01, Student’s t-test. (D) The luciferase reporter gene assay showed that miR-106b-5p mimic significantly decreased the luciferase activity of WT-LIF-3'-UTR, while MUT-LIF-3’-UTR was not affected. (E) When AR cells grew to about 70% confluence, transfected miR-106b-5p-mimic, antagomir, or their NC by Lipofectamine 2000. After 48 h, the expression of LIF was detected by Western blotting. n = 6. **p < 0.01, one-way ANOVA with Tukey’s correction.
LIF promoted cell apoptosis and inflammation of AR by promoting the activity of the JAK1/STAT3 signaling pathway
Previous studies have shown that LIF activated the JAK-STAT3 pathway,
22
and the JAK1/STAT3 signaling pathway is closely related with inflammation. As shown in Figure 5(A), the levels of LIF, p-JAK1, and p-STAT3 were up-regulated after IL-13 stimulation. After interfering the LIF, the levels of p-JAK1 and p-STAT3 were decreased. The apoptotic capacity of HNEpCs was weakened after interfering the expression of LIF (Figures 5(B) and (C)). In addition, knockdown of LIF decreased the expression of IgE, IL-4, and sVCAM-1 in IL-13 stimulated HNEpCs but up-regulated the expression of IL-10 (Figures 5(D)–(G)). Knocking down LIF up-regulated the expression levels of occludin and ZO-1 in HNEpCs treated with IL-13 (Supplementary Figure 3). In order to study the effect and mechanism of LIF on cell apoptosis and inflammation of AR, we used expression vectors to up-regulate the expression of LIF, siRNA to interfere the expression of LIF, and then detected the phosphorylation level of JAK1 and STAT3, cell apoptosis, and inflammation responses in the AR cell model. These data imply that LIF activated the JAK1/STAT3 signaling pathway to promote cell apoptosis and inflammation of AR (Supplementary Figure 4). LIF activated the JAK1/STAT3 signaling pathway to promote cell apoptosis and inflammation of AR. In IL-13 stimulated HNEpCs, siRNA was used to interfere the expression of LIF. (A) The expression of LIF, p-JAK1, and p-STAT3 was detected by Western blotting and quantized by Image J. (B) The proportion of apoptotic cells was detected by flow cytometry. (C) The expression of apoptosis-related proteins such as Bcl-2 and cleaved caspase-3 was detected by Western blotting and quantized by Image J. (D)–(G) The secretion of IgE, IL-4, IL-10, and sVCAM-1 in cell supernatant was detected by ELISA. n = 6. **p < 0.01, two-way ANOVA with Sidak’s correction.
LncRNA SNHG16 up-regulated LIF expression by binding with miR-106b-5p to promote cell apoptosis and inflammation of AR via the JAK1/STAT3 signaling pathway
In order to study the mechanism of SNHG16 affecting cell apoptosis and inflammation of AR, the shRNA of SNHG16 was transfected alone or together with miR-106b-5p antagomir into the AR cell model. As shown in Figures 6(A)–(D), after interfering the expression of SNHG16, LIF, p-JAK1, and p-STAT3 expression were down-regulated. And miR-106b-5p antagomir reversed the down-regulation of LIF expression and the inhibition of the JAK1/STAT3 signal pathway by SNHG16 shRNA. As shown in Figure 6(E), knockdown of SNHG16 decreased the apoptosis of AR cell model. In addition, Bcl-2 protein expression was up-regulated, and cleaved caspase-3 expression was down-regulated in the AR cell model with SNHG16 down-regulated. But miR-106b-5p antagomir reversed the inhibition of cell apoptosis (Figures 6(F)–(H)). Then we detected the expression of IgE, IL-4, IL-10, and sVCAM-1 in the cell supernatant by ELISA, and the results were shown in Figure 7. After interfering the SNHG16 expression, the secretion of IgE, IL-4, and sVCAM-1 was decreased, and the secretion of IL-10 was increased. MiR-106b-5p antagomir reversed the inhibition of AR inflammation. These results indicated that SNHG16 up-regulated the expression of LIF by binding with miR-106b-5p to promote the apoptosis and inflammatory response of AR cells via the JAK1/STAT3 signaling pathway. SNHG16 up-regulated the expression of LIF by binding with miR-106b-5p and promoted the apoptosis of AR cells via the JAK1/STAT3 signaling pathway. SNHG16 shRNA was transfected alone or together with miR-106b-5p antagomir into AR cell model. (A)–(D) The expression of LIF, p-JAK1, and p-STAT3 was detected by Western blotting, and quantitative analysis was performed using Image J. (E) Cell apoptosis was detected by flow cytometry. (F)–(H) The expression of apoptosis-related proteins such as Bcl-2 and cleaved caspase-3 was detected by Western blotting. n = 6. *p < 0.05, **p < 0.01, two-way ANOVA with Sidak’s correction. SNHG16 promoted the inflammatory response of AR cells. SNHG16 shRNA was transfected alone or together with miR-106b-5p antagomir into AR cell model: (A)–(D) The secretion of IgE, IL-4, IL-10, and sVCAM-1 in cell supernatant was detected by ELISA. n = 6. *p < 0.05, **p < 0.01, two-way ANOVA with Sidak’s correction.

Knockdown of SNHG16 reduced the inflammatory response of allergic rhinitis mice model
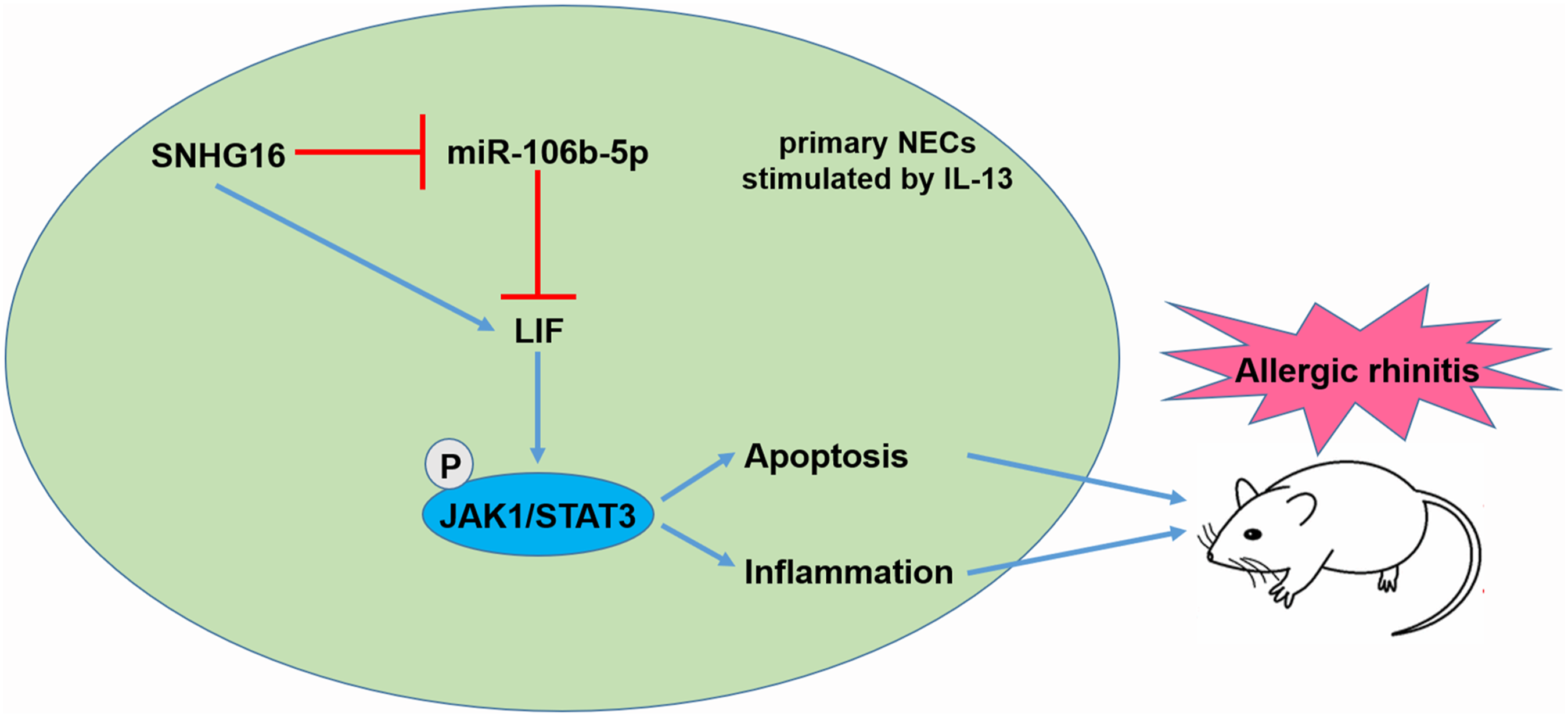
After successful modeling and treating, the histopathological changes of nasal mucosa were observed by HE staining. Compared with the control group, the rhinitis symptom score of the OVA group was significantly increased. The results of HE staining (Figure 8(A)) showed that the epithelium of nasal mucosa in the OVA group was exfoliated and infiltrated with a large number of inflammatory cells. After interfering the SNHG16 expression, a small amount of inflammatory cell infiltration could be seen. The expression of SNHG16 and LIF was up-regulated, the expression of miR-106b-5p was decreased in the OVA group (Figures 8(B)–(E)). The levels of p-JAK1 and p-STAT3 were up-regulated in the OVA group (Figures 8(D) and (F)). The secretion of IgE, IL-4, and sVCAM-1 in serum was significantly increased, and IL-10 was significantly decreased in the OVA group (Figures 8(G)–(J)). The interference of SNHG16 expression significantly reduced the rhinitis symptoms of AR mice, and inhibited the secretion of IgE, IL-4, and sVCAM-1, which might be related to the influence of miR-106b-5p and LIF expression, and further affected the activity of JAK1/STAT3 signaling pathway. A schematic diagram of the mechanism of SNHG16 function in AR pathogenesis was shown in Figure 9. In AR, the expression of lncRNA SNHG16 was up-regulated, and SNHG16 up-regulated the expression of LIF by combining with miR-106b-5p, thereby activating the JAK1/STAT3 signaling pathway and promoting the apoptosis and inflammation of AR cells. Knockdown of SNHG16 expression could relieve the rhinitis symptoms of AR mice. SNHG16 promoted inflammation of AR by binding with miR-106b-5p to promote the expression of LIF, thereby increasing the activity of the JAK1/STAT3 signaling pathway in vivo. Interfering the expression of SNHG16 in AR mice model, (A) the histopathological changes of nasal mucosa were observed by HE staining. (B) RT-qPCR was used to detect the interference effect. (C) The expression of miR-106b-5p was detected by RT-qPCR. (D)–(F) Western blotting was used to detect the expression of LIF and pathway-related proteins. The gray value was quantitatively analyzed by Image J. (G)–(J) The secretion of IgE, inflammatory factor IL-4, anti-inflammatory factor IL-10, and cell adhesion factor sVCAM-1 in cell supernatant was detected by ELISA. n = 10. **p < 0.01, two-way ANOVA with Sidak’s correction. A schematic diagram of the mechanism of SNHG16 function in AR pathogenesis.
Discussion
It was reported that in glioma, lncRNA SNHG16 promoted cell proliferation and inhibited cell apoptosis by inhibiting p21. 23 Other studies have shown that SNHG16 promoted the activation of PI3K/Akt signaling pathway via the miR-4518/PRMT5 regulatory axis to regulate the viability and apoptosis of glioma cells. 24 In osteosarcoma, down-regulation of SNHG16 inhibited cell proliferation, migration, and invasion but promoted cell apoptosis. 25 In the inflammatory response, it was found that SNHG16 up-regulated the inflammatory pathway, which was down-regulated by miR-15a/16 cluster. 26 It is reported that the expression of SNHG16 was up-regulated in the serum of patients with pneumonia. Down-regulation of SNHG16 significantly increased cell viability and inhibited cell apoptosis and the production of inflammatory cytokines via the miR-146a-5p/CCL5 axis. 27 These results suggested that SNHG16 affected cell apoptosis and the release of inflammatory factors in inflammatory response. Our study found that the expression of lncRNA SNHG16 was up-regulated in AR. And knockdown of SNHG16 could inhibit cell apoptosis and inflammation.
MiRNAs play important roles in various physiological and pathological processes, such as cancer progression 28 and inflammation.29,30 Studies have found that miR-106a inhibited oxidative stress injury and inflammatory infiltration in the liver of the mouse with gestational hypertension by inhibiting the mitogen-activated protein kinase signaling pathway. 31 MiR-106b expression was significantly inhibited in activated bone marrow derived dendritic cells (BMDCs) stimulated by OVA. And miR-106b negatively regulated the allergic properties of BMDCs and subsequent Th2 polarization by targeting early growth response (Egr)-2. 32 Here, we predicted and verified the binding of SNHG16 to miR-106b-5p by bioinformatics analysis, luciferase reporter gene analysis, and RNA pull-down experiment.
Leukemia inhibitory factor is the most effective cytokine in the interleukin-6 family. 33 LIF has different regulatory effects on cell apoptosis in different life activities. The expression of pro-apoptotic genes in human vitrified ovarian tissue of LIF-treated group was significantly lower than that of non–LIF-treated group. 34 However, studies have shown that miR-181a-5p inhibited apoptosis of pancreatic acinar cells by targeting LIF. 17 It was reported that IL-6 promoted inflammatory response in patients with inflammatory bowel disease by Th17 cell differentiation, which was induced by the activation of STAT3. However, LIF, an IL-6 family cytokine, inhibited inflammation by blocking the differentiation of Th17 cells. 18 But studies have shown that in LIF-deficient mice, neuroinflammatory responses were decreased. 35 LIF was overexpressed in arthritis. In a murine model of arthritis, compared with the wild-type disease control group, the clinical arthritis severity of LIF-deficient mice was reduced. 36 It was reported that LIF activated the JAK-STAT3 pathway by inducing heterodimerization of LIF receptor and the signal transduction protein gp130. 22 And the JAK1/STAT3 signaling pathway is closely related to inflammation. These results suggested that LIF may play a pro-inflammatory or anti-inflammatory role in different diseases. In this study, we demonstrated that miR-106b-5p directly targeted LIF, which promoted cell apoptosis and inflammation of AR by promoting the activity of the JAK1/STAT3 signaling pathway. SNHG16 could be used as an early diagnostic indicator for AR. Inhibiting the expression of SNHG16 could up-regulate the expression of miR-106b-5p and down-regulate the expression of LIF. And it could effectively alleviate the inflammation and damage of AR. These data provided a new theoretical basis for the clinical diagnosis and treatment of AR.
Conclusion
In this study, we found that knockdown of SNHG16 effectively ameliorates the pathology of AR through inhibiting cell apoptosis and reducing the inflammatory responses, which includes the concentration of IgE, IL-4, IL-10, and sVCAM-1. Notably, we observed that lncRNA SNHG16 directly binds with miR-106b-5p and subsequently up-regulates the expression of LIF, a target gene of miR-106b-5p. LIF activates the JAK1/STAT3 signaling pathway to promote cell apoptosis and inflammation of AR. These findings suggested that knockdown of SNHG16 may be a potential therapeutic approach for the treatment of AR.
Supplemental Material
sj-pdf-1-het-10.1177_09603271211035665 – Supplemental Material for Long non-coding RNA SNHG16, binding with miR-106b-5p, promoted cell apoptosis and inflammation in allergic rhinitis by up-regulating leukemia inhibitory factor to activate the JAK1/STAT3 signaling pathway
Supplemental Material, sj-pdf-1-het-10.1177_09603271211035665 for Long non-coding RNA SNHG16, binding with miR-106b-5p, promoted cell apoptosis and inflammation in allergic rhinitis by up-regulating leukemia inhibitory factor to activate the JAK1/STAT3 signaling pathway by Huajing Li, Fang Quan, Pengfei Zhang and Yuan Shao in Human & Experimental Toxicology
Supplemental Material
sj-pptx-2-het-10.1177_09603271211035665 – Supplemental Material for Long non-coding RNA SNHG16, binding with miR-106b-5p, promoted cell apoptosis and inflammation in allergic rhinitis by up-regulating leukemia inhibitory factor to activate the JAK1/STAT3 signaling pathway
Supplemental Material, sj-pptx-2-het-10.1177_09603271211035665 for Long non-coding RNA SNHG16, binding with miR-106b-5p, promoted cell apoptosis and inflammation in allergic rhinitis by up-regulating leukemia inhibitory factor to activate the JAK1/STAT3 signaling pathway by Huajing Li, Fang Quan, Pengfei Zhang and Yuan Shao in Human & Experimental Toxicology
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Science and Technology plan of Shaanxi Province-Key Research and Development plan (grant number 2020-SF-A20) and the Basic Research plan of Natural Science in Shaanxi Province (grant number 2020JM-382).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.