
Abstract
Acetaminophen (APAP) is often used as an antipyretic and analgesic agent. Overdose hepatotoxicity, which often results in liver cell failure and liver transplantation, is a severe complication of APAP usage. To save the liver and save lives from acute liver damage caused by APAP, the search for new strategies for liver defense is important. Wistar rats have been used for the induction of APAP hepatotoxicity. Elevated levels of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP) and lactate dehydrogenase (LDH) were evaluated for liver toxicity. In addition, the levels of hepatic tissue oxidative markers such as malondialdehyde (MDA), nitric oxide (NO) increased while glutathione (GSH) was depleted and catalase (CAT) activity was curtailed. The biochemical findings were consistent with the changes in histology that suggested liver damage and inflammation. Treated rats with N-acetylcysteine (N-AC) and granulocyte colony stimulating factor (G-CSF) showed a decrease in serum levels of ALT, AST and LDH, while the level of ALP in the G-CSF group was still high. After administration of APAP, treatment with N-AC or G-CSF substantially reduced the level of MDA and NO while maintaining the GSH content and CAT activity. Treatment with N-AC and G-CSF after administration of APAP has also attenuated inflammation and hepatocytes necrosis. The results of this study showed that G-CSF could be viewed as an alternative hepatoprotective agent against APAP-induced acute liver injury compared to N-AC.
Introduction
Due to severe side effects caused by oxidative stress with increased morbidity and mortality among patients, overuse of non-steroidal anti-inflammatory drugs (NSAIDs) is considered to be a health problem.1,2 Acetaminophen (N-acetyl-para-aminophenol, paracetamol, APAP) is an over-the-counter medication that produces safe and effective analgesic and antipyretic effects when used at therapeutic doses. 3 An acute or cumulative overdose of APAP could result in a major liver injury with acute liver injury (ALF). In developed countries, epidemiologically, APAP is the most common cause of ALF.4,5
APAP toxicity has been shown to consist of many steps and signaling pathways, including metabolism of APAP, oxidative stress, endoplasmic reticulum (ER) stress, autophagy, aseptic inflammation, microcirculation impairment, and compensatory repair and regeneration of the liver. 6 APAP overdose contributes to cellular protein conformation and functional changes, increased release of reactive oxygen (ROS) and reactive nitrogen (RNS) species, lipid oxidation, mitochondrial permeability and transitional pore opening. These are the significant events that lead to the death of cells.7–11
APAP metabolism takes place primarily in the liver. Glucuronidation and sulphation reactions are the primary mechanisms of APAP removal at therapeutic doses. However, high doses of the medication saturate the conjugation pathways with conversion of part of the drug to the highly reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) reacting with sulfhydryl groups. 12 NAPQI is initially trapped by GSH and the GSH adduct is excreted. NAPQI forms adducts by reaction with cellular proteins, including a variety of mitochondrial proteins in the event of GSH depletion. Consequently, this approach prevents mitochondrial respiration with the degradation of adenosine triphosphate (ATP) and mitochondrial oxidative stress.13,14
Previous incidents have increased vulnerability to ROS-related liver damage, including hydrogen peroxide (H2O2), superoxide anions (O2•-) and hydroxyl radicals (OH), as well as reduced levels of GSH. APAP overdose decreases the function of the antioxidant enzyme as CAT, increases lipid peroxidation and induces fragmentation of hepatic DNA, which eventually leads to cellular necrosis.15,16 Oxidative stress and mitochondrial dysfunction caused by APAP typically play a central role in the pathogenesis of acute liver failure. 17
The current treatment of choice for APAP overdose is N-acetyl-L-cysteine, a precursor of intracellular cysteine and GSH, which counteracts GSH depletion and enables NAPQI to be excreted as a GSH adduct. This decreases oxidative stress and, consequently, liver injury. 18 This drug also has its drawbacks, including side effects and narrow therapeutic window. 19 In patients with APAP-induced acute liver injury (AILI), liver transplantation is the only option to improve survival if the early and/or most treatable stage is skipped. 5
Recently, G-CSF therapy has resulted in promising outcomes in people with liver failure, this effect can be due to its ability to increase bone marrow stem cell production.20–22 Earlier studies have concluded that G-CSF attenuates liver injury and plays an important role in biology. This was evidenced by the ability of G-CSF to boost liver proliferative capacity in rat models of acute liver injuries and clinical trials involving patients with fulminant hepatic failure and acute hepatitis. 23
In animal studies, when G-CSF was administered to rats with liver damage, it promoted liver repair by increasing the migration of bone marrow progenitors to the liver, and by local action inside the liver microenvironment that facilitated the hepatic restoration program through oval cells. 24 In a model of liver damage, administration of G-CSF showed that it ameliorated the histological liver damage, accelerated the regeneration process and improved survival. 25 The beneficial effects of G-CSF on liver regeneration have also been shown in human trials. In a randomized controlled trial including patients with alcoholic steatohepatitis, G-CSF therapy mobilized CD34+ cells, increased hepatocyte growth factor, and induced hepatic progenitor cells to proliferate. 26
We investigated, in the present study, whether G-CSF prevents hepatocellular necrosis caused by APAP. In addition, we examined the extent of oxidative parameters and inflammatory infiltration in the rat liver after APAP administration to elucidate the underlying protective mechanism.
Materials and methods
Chemicals
Acetaminophen (APAP) (Sigma-Aldrich, Germany) was dissolved in 0.5% (w/v) methylcellulose (Sigma-Aldrich, Germany) solution, which was administered to the animals by oral gavage; N-acetylcysteine (N-AC) (AK Scientific, Inc. USA) was dissolved in 0.5% (w/v) methylcellulose solution, which was administered to the animals orally; G-CSF solution (filgrastim) (Roche, Egypt) was administered to rats subcutaneously; Malondialdehyde bis(dimethylacetal) (Merck, Germany); Glutathione reduced (ICN BIOMEDICALS, France).
Animals and experimental design
A total of 28 female Wistar rats (180–220 g) were obtained from Animal house, Faculty of medicine, Assiut University. The animals were placed in a 12 h light-dark cycle at constant temperature and humidity, and had free access to water during the experiment period. This study was done following the guidelines for animal experiments. Animals were carefully kept under the standard animal house conditions. There a permission from the medical ethics committee for this work (Medical Ethics Committee, Assiut University, IRB no. 17300515).
Rats were divided into four groups, each group including seven rats. Group 1: received carboxymethylcellulose (0.5%) vehicle orally for 5 days; Group 2: received acetaminophen (2 g/kg orally once) 27 for acute liver injury, the toxic dose of acetaminophen was given to this group 24 hours before sacrificing of all animals at the end of experiment; Group 3: received oral N-acetylcysteine (200 mg/kg) daily 28 after administration of acetaminophen for 5 days; Group 4: animals received acetaminophen then given G-CSF (filgrastim) 50 μg/kg subcutaneously 29 for 5 consecutive days.
After 24 hours, rats that received acetaminophen (2 g/kg orally) were anesthetized with urethane (0.75 mg/kg) intraperitoneal, and after the loss of straightening reflex, animals were killed by cardiac puncture with collection of blood and liver samples. The serum obtained from centrifugation was used for determination of standard biochemical parameters. Liver homogenate for each rat was prepared from 0.5 g of liver tissues, which were homogenized in Tris-HCl sucrose buffered solution at 4°C, this was used for estimation of glutathione level. Another liver homogenate was prepared from 0.5 g of liver tissues, which were homogenized in phosphate buffered saline at 4°C, this was used for estimation of the parameters of oxidative stress. For the first, third and fourth groups, the same was done after the fifth day of treatment with obtaining blood samples and liver specimens.
Biochemical measurements in serum
Liver toxicity was assessed by measurement of the serum levels of ALT, AST, ALP and LDH using a commercial spectrophotometrical method (SPECTRUM Diagnostics, Egypt). The values represent the mean of measurements ± standard error. The serum level of total protein was measured by commercial kit (SPECTRUM Diagnostics, Egypt).
Hepatic tissue MDA and NO measurement
Malondialdehyde is an end product of lipid peroxidation and it was determined by use of thiobarbituric acid reactive substance, method previously described by Ohkawa et al. 30 Malondialdehyde forms a 1:2 adduct with thiobarbituric acid, which can be measured spectrophotometrically at a wavelength of 532 nm.
Nitric oxide levels were measured using the Griess reaction31,32 which relied on a two-step process. In the first step, nitrates are converted to nitrites by nitrate reduction. In a second step, nitrite reacts with Griess reagent. At the end of this reaction, a dark purple azo compound is formed. The absorption of this azo compound was photometrically measured at a wavelength of 540 nm. The azo chromophore accurately determines nitrite concentrations as a marker of NO.
Hepatic GSH level and CAT activity measurement
The sulfhydryl group of GSH will react with 5,5 dithiobis-2 nitrobenzoic acid (DTNB, Ellman’s reagent) to produces a yellow colored 5-thio-2-nitrobenzoic acid (TNB). 33 An accurate estimation of GSH in the sample was provided by measurement of the absorbance of TNB at 412 nm.
The Aebi method 34 has been used to measure catalase (CAT) activity in liver tissue. H2O2 degradation was measured in the presence of catalase (CAT) at 240 nm. CAT activity was defined as the amount of enzyme required to process 1 nM H2O2 per minute at 26_C and pH 7.8.
Histopathology
For histopathological assay and to study the effect of APAP, G-CSF and N-AC on liver, samples were fixed in 10% (w/v) formalin for 24 h. After processing and embedding in paraffin using standard protocol, sections with 5 µm thickness were stained with hematoxylin and eosin to evaluate liver damage. For quantitative evaluation, histological photomicrographs were evaluated by scoring system. According to the extent of lobular distortion, necrotic changes and inflammatory cell infiltrations and congestion were scored as 0 (normal), 1 (mild), 2 (moderate), or 3 (severe). 35
Statistical analysis
Statistical analysis was performed using GraphPad Prism seven software (GraphPad Software). Normally distributed data was tested by one-way ANOVA with a posthoc Tukey’s correction. In all tests a 95% confidence interval was used, for which p < 0.05 was considered a significant difference. All data are represented as mean ± S.E. 36
Results
Serum levels of ALT, AST, ALP, LDH and total protein
Administration of acetaminophen orally in a dose of 2 g/kg to rats resulted in significant elevation (p < 0.01) of the serum levels of ALT (40.50 ± 1.80 U/L), AST (159.7 ± 9.25 U/L), ALP (455.5 ± 26.81 U/L) and LDH (646.6 ± 23.79 U/L) when compared to control rats that received the vehicle [ALT (21.83 ± 2.072 U/L), AST (70.83 ± 2.822 U/L), ALP (206.0 ± 20.72 U/L) and LDH (173 ± 9.29 U/L)]. Treatment with N-AC and G-CSF produced a significant reduction (p < 0.01) in the serum level of these biochemical parameters compared to APAP-treated rats except the ALP level in G-CSF-treated rats as It was noticed that G-CSF administration after APAP produced an elevation of the serum level of ALP. For N-AC group [ALT (29.00 ± 1.983 U/L), AST (110.8 ± 9.057 U/L), ALP (287.0 ± 23.54 U/L) and LDH (206.3 ± 7.637 U/L)] and for G-CSF group [ALT (26.33 ± 1.745 U/L), AST (91.67 ± 6.581 U/L), ALP (532.8 ± 29.01 U/L) and LDH (239.4 ± 13.07 U/L)] (Figure 1(a), (b), (c) and (d) respectively). Also, APAP administration resulted in a decrease in the serum level of total protein (4.04 ± 0.21 g/dL) (p < 0.01) compared to control rats (6.117 ± 0.29 g/dL) and this reduction was alleviated in rats that treated with N-AC (5.687 ± 0.2248 g/dL) and G-CSF (5.634 ± 0.1985 g/dL) (p < 0.01) (Figure 2).
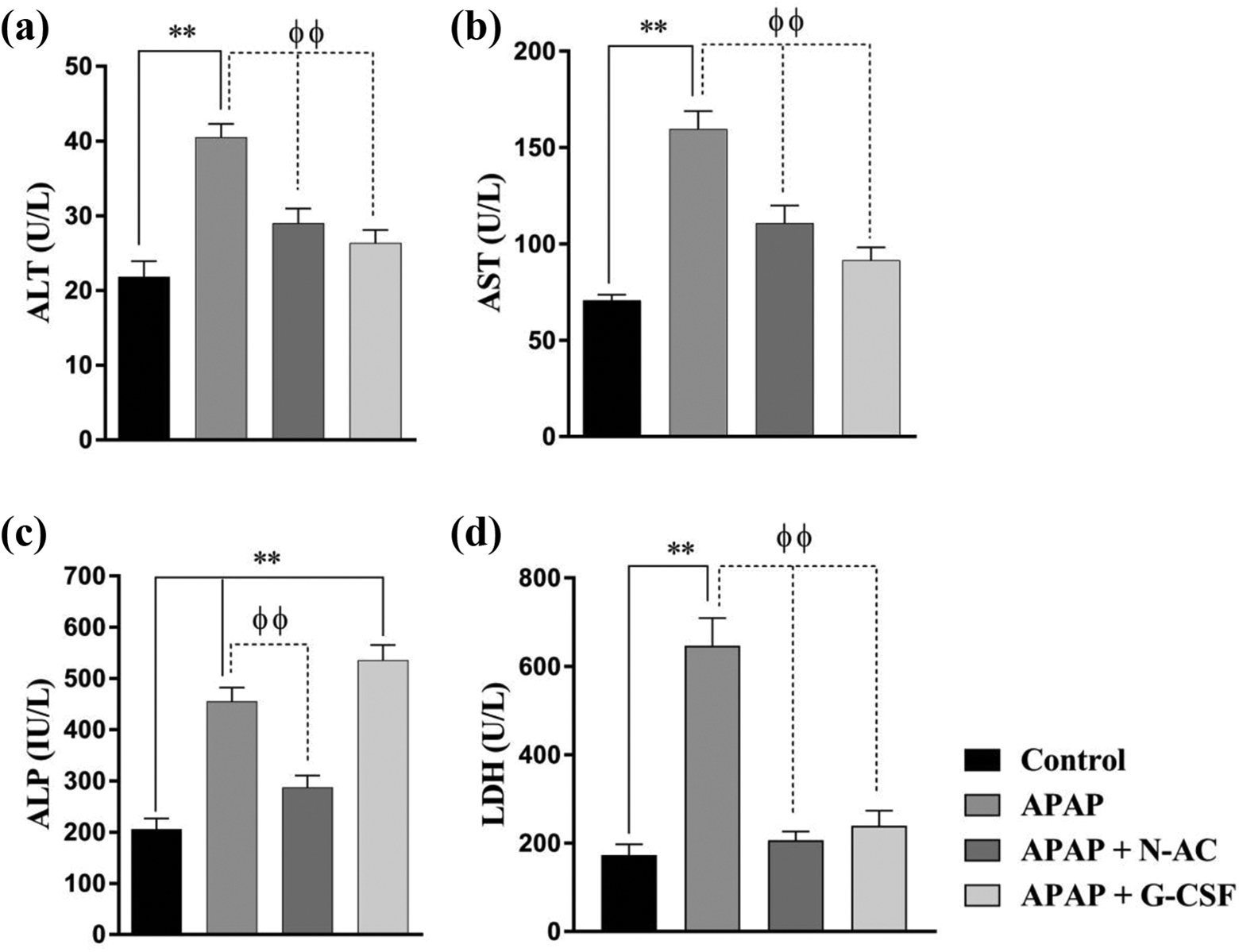
Effect of APAP, N-AC and G-CSF on the serum level of ALT (a), AST (b), ALP (c) and LDH (d). Data presented as mean ± SEM of seven observations. **Significance at p < 0.01 vs. control animals. φφSignificance at p < 0.01 vs. APAP-received animals.

Effect of APAP, N-AC and G-CSF on the serum level of total protein. Data presented as mean ± SEM of seven observations. **Significance at p < 0.01 vs. control animals. φφSignificance at p < 0.01 vs. APAP-received animals.
Liver tissue MDA and NO levels
As seen in Figure 3((a) and (b)) Acute intoxication by oral administration of APAP (2 g/kg) significantly enhanced the MDA (1191 ± 70.50 nmol/g.w.wt, p < 0.01) and NO (12 ± 0.6901 μmol/g protein, p < 0.01) levels in liver tissue homogenate in comparison to control group with MDA (476.2 ± 47.67 nmol/g.w.wt) and NO (5.33 ± 0.38 μmol/g protein) . Therapy with N-AC after administration of APAP for 5 consecutive days significantly reduced MDA (509.7 ± 62.76 nmol/g.w.wt, p < 0.01) level and NO (5.586 ± 0.3203 μmol/g protein, p < 0.01) level. As well, G-CSF administration produced similar effects with lowering of MDA level (503 ± 34.40 nmol/g.w.wt, p < 0.01) and NO level (5.886 ± 0.3522 μmol/g protein p < 0.01).

Effect of APAP, N-AC and G-CSF on the hepatic level of MDA (a) and NO (b). Data presented as mean ± SEM of seven observations. **Significance at p < 0.01 vs. control animals. φφSignificance at p < 0.01 vs. APAP-received animals.
Liver tissue GSH level and CAT activity
APAP administration declined GSH content in hepatic tissue (1.836 ± 0.19 µmol/g.w.wt, p < 0.01) in comparison to control rats (6.037 ± 0.87 µmol/g.w.wt). This GSH depletion was corrected by treatment with N-AC (4.954 ± 0.5 µmol/g.w.wt, p < 0.01) and G-CSF (5.617 ± 0.49 µmol/g.w.wt, p < 0.01) (Figure 4(a)). Concomitantly, rat livers that received APAP showed a significant low CAT activity (10.5 ± 0.69 mmol/min/mg tissue, p < 0.01) compared to control rats (21.43 ± 0.87 mmol/min/mg tissue). N-AC treatment corrected this low CAT activity (18.16 ± 0.51 mmol/min/mg tissue, p < 0.01) as well as treatment with G-CSF (18.21 ± 0.63 mmol/min/mg tissue, p < 0.01) (Figure 4(a)).
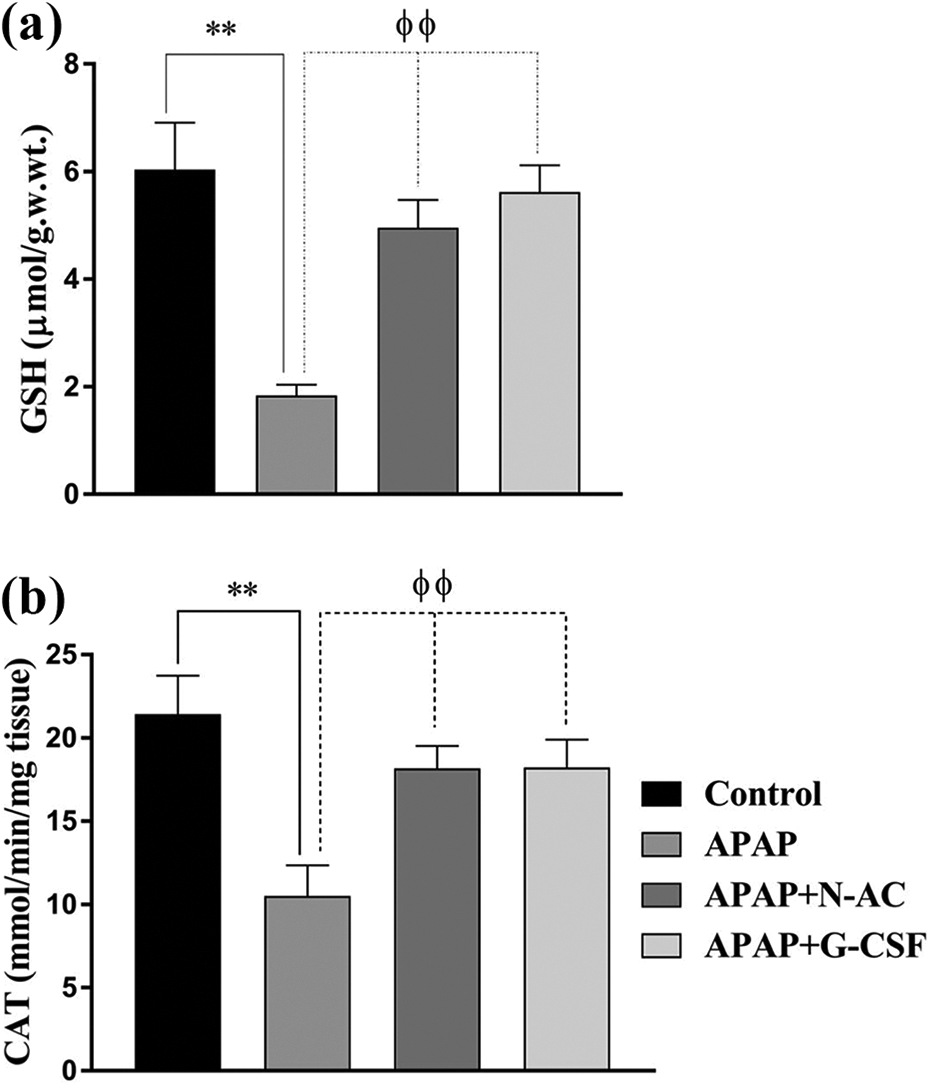
Effect of APAP, N-AC and G-CSF on the hepatic level of GSH (a) and CAT activity (b). Data presented as mean ± SEM of seven observations. **Significance at p < 0.01 vs. control animals. φφSignificance at p < 0.01 vs. APAP-received animals.
Histopathological examination of liver tissue
Liver tissue stained with hematoxylin and eosin and examined under light microscope showed the presence of necrosis, inflammatory cell infiltration with loss of normal hepatic architecture in rats treated with APAP compared to control rats (Figure 5(a)–(d)). Treatment with N-AC and G-CSF after APAP administration for 5 consecutive days resulted in reduction of pathological finding in liver tissue indicated by amelioration of inflammatory cell infiltration and restoration of hepatic microanatomical features when compared to rats with APAP-induced hepatic damage (Figure 5(e)–(g)). Liver injury score in Figure 6 indicated the protective effect of G-CSF against APAP-induced hepatotoxicity which was comparable to the effects of N-AC.

((a) and (b)) represent the control group, Scale bar: 100 and 20 μm. Effect of G-CSF and N-AC on APAP-induced acute liver injury determined with histological H & E staining. Tissue section photography ((c) and (d)) represent a lobular distortion due to the presence of wide necrotic patches which characterized by complete loss of cellular details (notched arrow) in APAP group, Scale bar: 100 and 20 μm. ((e) and (f)) N-AC+ APAP, ((g) and (h)) G-CSF + APAP represent a mild inflammatory response and congestion with nearly normal lobular architecture, Scale bar: 100 and 20 μm.
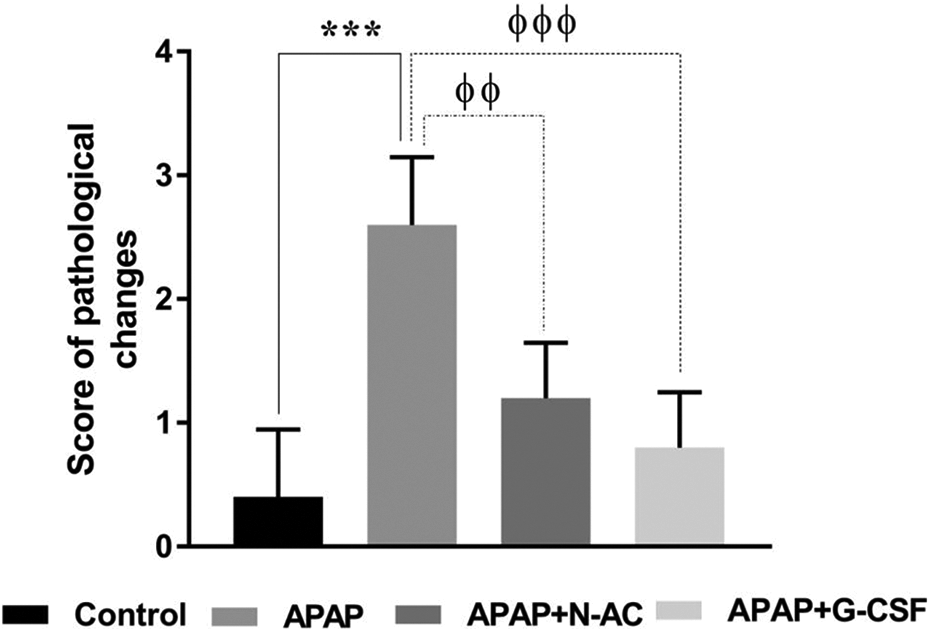
Histogram shows Liver injury score in liver tissue. Data presented as mean ± SEM. ***p < 0.001 vs control group; φφp < 0.01 vs the APAP groups; φφφp < 0.001 vs APAP group.
Discussion
Hepatotoxicity caused by acetaminophen is one of the medical conditions requiring the pursuit for alternative treatment strategies rather than conventional treatment lines. G-CSF has been shown to mitigate ALT activities, enhance liver regeneration and improve survival in the event of liver injury. In line with this, this experimental research was planned to test the effects of G-CSF in APAP-induced liver injury, as it is a common form of drug-induced hepatotoxicity.
In this study, the hepatotoxic toxic effect of APAP is determined by a substantial increase in the serum levels of AST, ALT, ALP and LDH in APAP-treated rats, as compared to control group. Azim et al. 37 found that rats treated with toxic doses of APAP showed the same results of our study regarding liver enzymes. These ALT, AST, ALP and LDH activities are used as biochemical indicators for early hepatic injury assessments. Arvind Kumar and Sangeeta 38 also showed that exposure to APAP in rats significantly increased the serum levels of AST, ALT, LDH and ALP. After cellular leakage and loss of functional integrity of the cell membrane in the liver, these enzymes are released into the bloodstream due to hepatotoxicity. 39 Elevated serum AST, ALT ALP and LDH activities are indicative of cellular leakage and loss of functional integrity of the hepatocellular membrane.40,41
A possible explanation by which serum ALP level was elevated may be related to the increase in its synthesis by cells lining bile canaliculi in response to cholestasis and increased biliary pressure. The aforementioned increase of liver parameters in APAP-rats is also supported by preceding studies that agreed with our findings. In addition, APAP decreased serum total protein level in the present study, as well, reported by Lotková et al. 42 who demonstrated a significant decrease in ALB synthesis in APAP-treated group in comparison to controls.
The findings of the present study demonstrated that APAP overdose induced an imbalance between ROS production and antioxidant protection, resulting in lipid peroxidation as evidenced by the increased hepatic contents of MDA and NO while the decreased level of GSH and diminished CAT activity. Previous studies agree with our findings regarding the levels of MDA, NO, GSH and CAT activity which indicated hepatic oxidative stress induced by APAP toxicity.1,43,44 The destructive process of the liver due to APAP intoxication has been reported to be lipid peroxidation. 45 The rise in thiobarbituric acid reactants (TBARS) called for an enhancement of lipid peroxidation (LPO), leading to tissue damage and failure of the antioxidant defense mechanism to prevent the excessive formation of free radicals. APAP’s highly reactive metabolite metabolism, NAPQI, and its GSH detoxification are serious determinants of APAP’s toxicity. 46
Furthermore, hepatocellular necrosis induced by APAP is exacerbated by the massive activation of inflammatory reactions caused by the development of cytokines and chemokines by Kupffer cells in response to hepatocellular necrosis and infiltration of inflammatory cells. These results were confirmed by histopathological modifications in our specimens. In APAP-induced hepatotoxic rat models, several studies found the same findings.47–49
Our research emphasizes the therapeutic function of N-AC in hepatotoxicity induced by APAP; we found that N-AC administration to rats received APAP provided attenuation of elevated serum AST, ALT, ALP and LDH for several days. In addition, levels of the oxidative stress parameter in hepatic tissue have decreased significantly. Histopathological abnormalities observed in rats treated with APAP were alleviated with N-AC administration. Many studies support the role of N-AC in hepatotoxicity induced by APAP.50–54
When administered at an early stage of toxicity, N-acetyl cysteine, a well-known ROS scavenger, has been accepted as an important therapeutic antidote for acute APAP-induced liver injury (AILI). The narrow treatment window, unfortunately, restricts its use. The need for new therapeutic methods to produce protective effects against AILI is therefore mandatory.
In an experimental model, G-CSF was administered immediately after liver intoxication with a single intraperitoneal dose of carbon tetrachloride (CCl4), resulted in a decrease in the activity of liver enzymes, ALT, AST and LDH compared to the untreated group. 55 In contrast, a study did not support a protective effect of G-CSF against toxic, non-lethal acute liver injury and promoted hepatocyte regeneration. 56 Liu et al. 57 found that increased levels of circulating lipopolysaccharide (LPS) contributed to an imbalanced inflammatory response and microcirculatory dysfunction that preceded liver damage and subsequent dysfunction. Pre-treatment with G-CSF exacerbated microcirculation disturbances and liver injury. This may be related to G-CSF-induced LPS sensitization.
We found that G-CSF administration in rats with APAP-induced hepatotoxicity has been shown to improve the profile of liver enzymes and oxidative stress parameters, except for ALP, which is still elevated in the blood. The elevated level of ALP is a matter of debate and multiple explanations were postulated. In mice, Wang et al. 58 concluded that G-CSF had preventive and treatment effects on CCl4 induced chronic liver injury. It is suggested that G-CSF increased the survival rate, decreased liver injury and enhanced hepatocyte proliferation in rats with D-galactosamine-induced acute liver failure, this was possibly through actions including but not limiting to CD34+ cell mobilization, and that G-CSF may be of potential value in treating ALF. 21
In addition, the influence of G-CSF on the proliferative response of progenitor cells in the bone and liver can explain this unexpected increase in serum ALP levels. This effect was observed in other studies involving the administration of G-CSF with a substantial increase in serum ALP levels. 59
Nam et al. 60 found that hepatotropic dose of G-CSF significantly reduced hepatocyte apoptosis via PI3 K and Akt pathway activation without marrow cell mobilization in non-alcoholic fatty liver disease animal model. Preliminary findings further underline the complexity by which G-CSF might modulate sterile inflammation and liver regeneration in patients with liver failure. Monitoring peripheral stem and immune cell levels might help to define subgroups of patients who benefit most from G-CSF administration. Engelmann and his colleagues have initiated a prospective randomized multicenter trial aiming at further elucidation the role of G-CSF in patients with acute-on-chronic liver failure (ACLF). A clinical sub-study would elucidate the importance of stem and neutrophil levels and functions in ACLF patients treated with G-CSF. 61 The aim of these clinical studies is to illustrate the underlying mechanisms of G-CSF in acute liver failure patients. There are some suggested explanations including proinflammatory cells attenuation, immune response modulation reduction of injurious free radicals release and triggering liver cell regeneration.
Both animal and human evidences suggest that stem cell translocation from the bone marrow to the liver is biologically possible, giving rise to cytokine therapies based on stimulation of the bone marrow. The most widely used cytokine for this function has been the Granulocyte colony stimulating factor. In small clinical trials, this intervention has shown promising results in terms of protection as well as survival benefits. However, the proof is incomplete and heterogeneous. 62
We found that G-CSF could improve liver function and cellular regeneration in necrotic liver injury induced by APAP, histological results suggest that evidence of regeneration and reduction of apoptotic markers can support the beneficial role of this important factor in hepatoprotection either in acute and chronic liver injuries.
We propose that, through various mechanisms, G-CSF may protect the liver (Figure 7). It has the ability to initiate bone marrow cellular migration and hepatic progenitor cell stimulation. In addition, the expression of growth factors within and outside the liver may be increased. The development of anti-inflammatory cytokines and antioxidant enzymes with attenuation of proinflammatory cytokines and ROS could be initiated by G-CSF. These suggestions agree with other studies regarding the way the drug works in protecting the liver from various diseases, including hepatitis C infections 63 and severe alcoholic hepatitis. 64

An illustration of the possible mechanisms of G-CSF in APAP-induced hepatotoxicity.
We can conclude that G-CSF and N-AC produced a comparable effect as therapeutic tools for APAP-induced liver injury; G-CSF has a specific guidance in administration regarding its dose. It requires further evidence from clinical studies to establish the therapeutic role of G-CSF in APAP hepatotoxicity and to improve the survival rate after exposure and decrease requirements for transplantation.
Footnotes
Acknowledgment
We thank the medical and technical staff of the Pharmacology department, Faculty of Medicine, Assiut University. Many thanks to all that help us to perform this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.