
Abstract
Introduction:
Di-(2-ethylhexyl) phthalate (DEHP) is a commonly used plasticizer in consumer products and medical devices. It is also suspected to exacerbate the development of fatty liver. However, the mechanisms underlying excessive lipid synthesis and its deposition in the liver are yet to be identified. This study was aimed to evaluate the molecular mechanisms of hepatic lipid accumulation in adult male offspring after perinatal exposure to DEHP.
Method:
Corn oil and DEHP (0.75 mg/kg/day) were administered once per day to dam from gestation day 6 to postnatal day (PND) 21 by oral gavage. After the weaning period, DEHP treated male pups were categorized into early life stage- and lifelong period group. Male rats both control and early life stage group administered corn oil, and lifelong period group administered DEHP from PND 22 to 70. Histological examination and triglyceride (TG) levels in the liver were analyzed. Expressions of transcription factors associated with lipid accumulation in the liver were analyzed.
Results:
Both early life stage- and lifelong period group, hepatic TG levels, and mRNA and protein expression of diacylglycerol acyltransferase 1 (DGAT1) were significantly higher than control (TG: all p < 0.05, mRNA & protein: p < 0.05 and p < 0.001, respectively). The average body weight from PND 35 to 63, and mRNA and protein expression of sterol regulatory element binding protein 1c in lifelong period group were significantly lower than control (all p < 0.05); however, alanine transaminase were significantly higher than control (p < 0.01).
Conclusion:
Perinatal exposure to DEHP may induce the hepatic lipid accumulation through up-regulation of DGAT1 expression.
Keywords
Introduction
Di-(2-ethylhexyl) phthalate (DEHP) is the most widely used additive for increasing the flexibility of plastic-based consumer products and medical devices. Approximately two million tons of DEHP are produced annually worldwide. 1 With the widely usages, the public concerns are growing of phthalate exposure and their impacts on human health. According to epidemiological and toxicological studies, exposure to DEHP may be associated with obesity, diabetes mellitus, and adipocyte accumulation.2–6 Recent studies have suggested that DEHP might cause increased adiposity or non-alcoholic fatty liver disease (NAFLD).7–10
NAFLD is the most common chronic liver disease and is closely associated with obesity, insulin resistance, type 2 diabetes mellitus, hypertension, hyperlipidemia. 11 It is characterized by a build-up of excess fat in over 5% of hepatocytes, without the involvement of significant alcohol intake and other known diseases of the liver. 12 It ranges from simple steatosis and fatty liver during early stages to non-alcoholic steatohepatitis (NASH), fibrosis, and cirrhosis in advance stages. 11 The prevalence of NAFLD is growing worldwide and is particularly high-fat diet consuming countries. 13
The liver plays a central role in maintaining glucose and insulin homeostasis, as well as in lipid metabolism. In general, free fatty acids released from adipose tissue are transferred to the liver and converted into triglycerides (TGs) which are the major storage molecules in the liver. Because excessive TG accumulation might induce obesity and other metabolic diseases, a better understanding of the process of TG synthesis and storage in the liver is required.
Synthesis of TGs is catalyzed by acyl-CoA: diacylglycerol acyltransferase (DGAT) enzymes, i.e. DGAT1 and DGAT2.14,15 When the expression of either enzyme is increased, synthesis and accumulation of TGs are consequently elevated. 16 Previously, both in vivo and in vitro studies have shown that DEHP induces the expression of genes related to β-oxidation and fatty acid metabolism in the liver.7–10 Despite those studies highlighting the relationship between DEHP and hepatic lipid accumulation, the molecular pathways induced upon exposure to DEHP still remain undetermined. In addition, it has not been studied whether early life exposure to DEHP can result in an increase of hepatic lipids in adult offspring.
Therefore, this study was performed to investigate the molecular mechanism of lipid accumulation in the liver of sub-adult male offspring after perinatal exposure to DEHP.
Materials and methods
Animals
Eight weeks of aged specific pathogen-free pregnant Sprague-Dawley rats (Rattus Norvegicus, n = 15, average weight 277.2 ± 27.7 g) were purchased from the Charles River Orient Experimental Animal Breeding Center (Gyeonggi-do, Korea). The female rats whose mating was confirmed with a vaginal plug were purchased. The day when mating was confirmed was set as gestational day (GD) 0. The rats were reared in stainless steel cage (215W×355L×200H mm) during the acclimatization period, quarantine period and mating period. After the accumulation, pregnant rats were identified and singly housed in polycarbonate cage (240L×390W×180H mm) with wood bedding to minimize additional exposure to endocrine-disrupting chemicals. They were maintained in a controlled environment (22 ± 2°C; 14 h light-10 h dark cycle, relative humidity at 55 ± 15%), and provided laboratory chow pellets (Purina Korea Inc., Gyeonggi-do, Korea) and UV-purified and filtered water ad libitum. Animal experiments were performed in the Korea Testing and Research Institute according to Good Laboratory Practice guidelines (TBH-308, 2010/07).
Dose selection
Dosing solutions were prepared by dissolving DEHP at 0.75 mg/kg/day in corn oil. The dosage amount was adjusted daily based on the body weight. The administered amount was calculated as 10ml/kg. The injection volume limited 2 ml in the rat. The dose level of DEHP that was used in this study was chosen on the basis of a previous study. 17 The levels of DEHP was approximately six times lower than No Observed Adverse Effect Levels (NOAEL) for reproductive and endocrine effects (5 mg/kg/day) reported by the European Food Safety Authority. 18
Experimental protocol
Pregnant rats were randomly divided into two groups, i.e. control (n = 5) and DEHP 0.75 mg/kg/day treated (n = 10) groups. The number male offspring on PND 0 were 21 in control and 36 in DEHP treated group. On PND 4, the number of male offspring were adjusted as 10 in control and 20 in DEHP treated group. Each group was administered corn oil or DEHP from gestational day (GD) 6 to postnatal day (PND) 21. After the weaning period, male offspring in DEHP treated group were divided into early life stage- and lifelong period groups (n = 8/group). Male offspring in control and early life stage group were administered corn oil, and lifelong period group received DEHP by gavage from PND 22 to PND 70. The details of administration schedule were described in our previous study. 19
At the end of our experiment (PND 69), the animals were fasted overnight with free access to water. Male offspring were sacrificed under isophorone anesthesia via inhalation on the morning of PND 70. After confirming the anesthesia state, the rat was taken out from the cage and blood was collected from the vena cava using needle (21 to 25G). The liver was weighed based on which relative liver weight was calculated (liver weight/body weight ×100, %). Part of the liver was collected and fixed in 10% neutral buffered formalin solution. The remaining livers were stored at −80°C until analyzing the effects of changes in mRNA and protein expression on lipogenesis.
Body weight gain
After spontaneous delivery, the body weight of male offspring was measured and recorded on PND 0, 4, 7, 14, and 21, and weekly thereafter until PND 70.
Food and Water intake
After the weaning period, the amount of food and water intake was measured and recorded at weekly intervals from PND 22 to PND 70.
Blood biochemical analysis
The collected blood was separated to serum and erythrocytes by centrifugation at 1000 ×g for 10 min at 4°C. Serum (500 μl) was drawn to sample cup to measure the total cholesterol (T-Cho), TG and liver enzymes such as aspartate aminotransferase (AST), and alanine transaminase (ALT). Sample cups were placed on the sample shelf of the automatic serum analyzer (Hitachi 7060, Tokyo, Japan).
Hepatic TG content
Liver samples were rinsed with ice-cold phosphate-buffered saline (PBS) to remove excess blood, and subsequently homogenized in PBS (1:9, w/v). Suspensions were sonicated to further break the cell membranes. After that, homogenates were centrifuged at 5000 ×g for 5 min. Supernatants were collected to measure hepatic TG. The preparation and measurement of TG in liver sample was in accordance with the manufacturer’s protocol (MyBiosource, Inc., San Diego, CA, U.S.A.). The absorbance (450 nm) was measured with a plate reader (Tecan Sunrise TW, Salzburg, Austria).
Histological examination of the liver
Liver samples fixed with formalin solution was dehydrated in graded ethanol for 24–48 hours and embedded in paraffin. Embedded tissue was sectioned into 4 µm thick slices and deparaffinized using xylene and 100–70% ethanol, and rehydrated using distilled water. Excess water in slides removed using a Kimwipe and stained with hematoxylin and eosin (H&E) for conventional morphological evaluation.
Stained slides were analyzed by light microscopy (Olympus, Tokyo, Japan). To examine the presence of steatosis in the liver, macro vesicular and micro vesicular were observed. The number of small and larger droplets were counted for 10 random fields for each rat liver at 400×. 20 The photographs in the present study were acquired with a Fuji Digital Camera HC300Z/CL (Olympus, Tokyo, Japan).
TG related genes and protein expression in the liver
Gene expression in the liver (qRT-PCR)
Total RNA was isolated from liver tissue using an RNA isolation kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Isolated total RNA was used to synthesize first-strand cDNA using PrimeScript™ RT Master Mix (TAKARA, Otsu/Shiga, Japan). Rat-specific primers were used to amplify the target genes. RT-PCR reactions were performed in duplicates using the Thermal cycler dice® real-time system (TAKARA, Otsu/Shiga, Japan). Expression was normalized to the average of rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH), β-actin, and ribosomal protein lateral stalk subunit P0 (RPLP0). Analysis of mRNA expression of target genes was performed by qPCR, followed by calculation employing the ΔΔCt method. The sequences of the primers used are listed in Table 1. RT-PCR reactions were carried out according to the following protocol: 1 cycle of initial denaturation at 95°C for 2 min, 45 cycles of 10 sec each at 95°C for denaturation, and 40 sec at 62°C for annealing and extension.
Sequences of primers used for real-time PCR amplification.
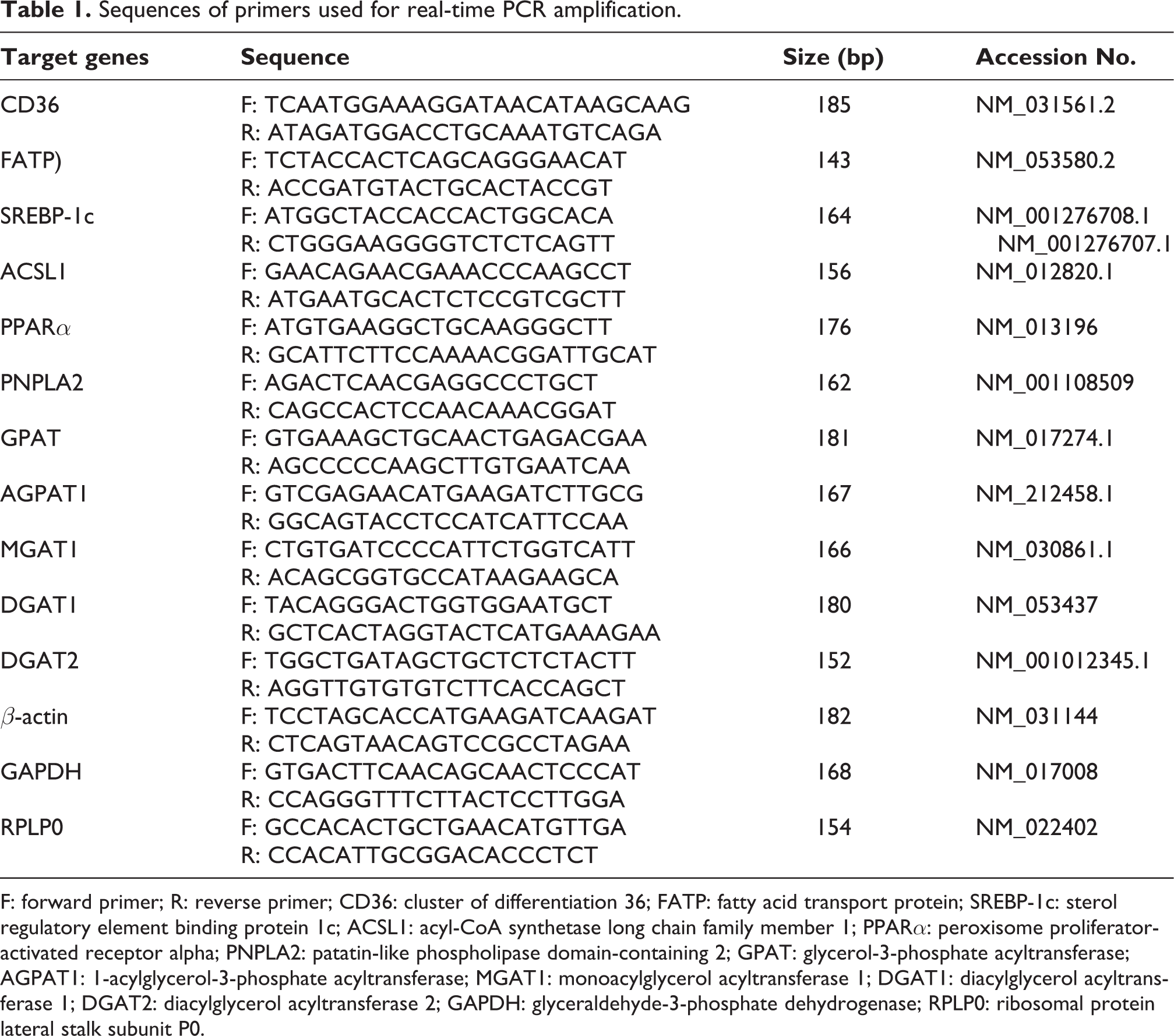
F: forward primer; R: reverse primer; CD36: cluster of differentiation 36; FATP: fatty acid transport protein; SREBP-1c: sterol regulatory element binding protein 1c; ACSL1: acyl-CoA synthetase long chain family member 1; PPARα: peroxisome proliferator-activated receptor alpha; PNPLA2: patatin-like phospholipase domain-containing 2; GPAT: glycerol-3-phosphate acyltransferase; AGPAT1: 1-acylglycerol-3-phosphate acyltransferase; MGAT1: monoacylglycerol acyltransferase 1; DGAT1: diacylglycerol acyltransferase 1; DGAT2: diacylglycerol acyltransferase 2; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; RPLP0: ribosomal protein lateral stalk subunit P0.
Protein expression in the liver (Western blot)
Liver samples were homogenized in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich Co. LLC., St Louis, MO, USA) to lyse the cells. Nuclei and intact cells in the liver samples were separated using centrifugation at 760 ×g for 10 min. The supernatant was transferred to a fresh tube and centrifuged at 12,000 ×g for 20 min. The resulting supernatant was collected and used for all subsequent protein assays (ThermoFisher Scientific Inc., Waltham, MA, U.S.A.). Aliquots (50 µg protein each) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (GE Healthcare Life Sciences, Pittsburg, PA, U.S.A.). These membranes were then blocked with 5% (w/v) skim milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) and incubated with primary antibodies directed against GAPDH (Santa Cruz Biotechnology Inc., Dallas, TX, U.S.A., sc-32233, 1:2000), DGAT1 (ThermoFisher Scientific Inc., Waltham, MA, U.S.A., PA5-18634, 1:1000) and sterol regulatory element binding protein 1c (SREBP-1c; Novus biologicals, Centennial, CO, U.S.A, NB100-2215, 1:500) in TBS-T. Following incubation, the membranes were washed three times with TBS-T for 10 min each, and then incubated with anti-rabbit IgG (Santa Cruz Biotechnology Inc. Dallas, TX, U.S.A., sc-2004, 1:4000) or anti-goat IgG respectively (Santa Cruz Biotechnology Inc. Dallas, TX, U.S.A., sc-2354, 1:4000) secondary antibodies, respectively. After washing the membrane, exposed films were scanned, and images were obtained using LAS 4000 instrument (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, U.S.A.). The density of each band was quantified using ImageQuant TL software (GE Healthcare Bio-Sciences Corp. Piscataway, NJ, U.S.A.).
Statistical analysis
Data on body and relative liver weights, liver TGs, as well as serum AST, ALT, T-Cho, TGs, and qRT-PCR and western blot are expressed as means ± standard error (SE). To test for normality, Shapiro-Wilk test was performed. Because of the outlier and small sample size, Kruskal-Wallis test was used for comparisons among groups. After wards, Dunnett’s test was performed to determine statistical significance of the observed differences. Data analysis was performed using STATA (version 16.0 StataCorp LP College Station, TX, USA). P-values ≤0.05 were considered a statistically significant.
Results
Body weight gain
No difference was observed in the average body weight from PND 0 to 21 (weaning period) between control- and DEHP-treated groups (Figure 1(a)). The early life stage group showed significantly higher body weights than those of the control group on PND 28 (p < 0.05; Figure 1(b)). Further, average body weight of the early life stage group was showed similar to that of the control group. In the lifelong period group, body weights were significantly lower than those of control rats on PND 35, 42, 49, 56, and 63 (all p < 0.05; Figure 1(b)). Body weights in the lifelong period group were significantly lower than in the early life stage group on PND 35, 63, and 69 (all p < 0.05; Figure 1(b)).

Effects of DEHP exposure on (a and b) body weight and (c) food intake in male offspring. (a) Average body weight in male pups before weaning period. (b) After the weaning period, DEHP treated group was divided into early life stage and lifelong period (n = 8 per group). Data represent the mean ± standard error. *p < 0.05 for early life- and lifelong period group vs control group; #p < 0.05, ##p < 0.01 for early life stage- vs lifelong period group.
Food and water intake
The difference in body weight gain among experimental groups was independent of the amount of food because there were no statistical significance of food intake levels (g) from PND 28 to 69 in early life stage and lifelong period groups compared to the control group (Figure 1(c)). The amount of water intake was no differ between groups from PND 28 to 69 (data not shown).
Relative liver weight and blood values
Relative liver weight (%) in the early life stage- and the lifelong period groups was similar to that in the control group (Table 2).
Relative liver weight and blood biochemical values in adult male offspring (n = 8/group).

T-Cho: Total cholesterol; TG: triglycerides; AST: aspartate aminotransferase; ALT: alanine transaminase.
* p < 0.05, ** p < 0.01 for early life- and lifelong period group vs control group.
# p < 0.05 for early life group vs lifelong period group.
Serum TG levels were significantly lower in the early life stage group than in control and lifelong period group (all p < 0.05; Table 2). Serum AST, ALT and T-Cho levels in early life stage group rats were similar to those of the control group. Serum levels of ALT in the lifelong period group showed were significantly elevated compared with those in the early life stage group (p < 0.01). Blood chemistry analyses on the lifelong period group did not reveal any statistically significant differences when compared with the control group.
Hepatic TG levels
Hepatic TG levels in early life stage and lifelong period groups were significantly higher than in the control group (all p < 0.05; Table 2). There was no significant difference between early life stage- and lifelong period group.
Histological findings in the liver
Hepatocytes, central veins, and portal areas in liver in the control, early life stage and lifelong period groups showed normal appearances (Figure 2(a)). Early life stage and lifelong period groups revealed few small lipid droplets localized within hepatocytes (Figure 2(a) middle and right panels). The number of macro lipid droplets in the liver tended to increase in the early life stage and lifelong period groups compared with control group; however, the difference was not significant (Figure 2(b)). A significant increase was observed in small lipid droplets in both early life stage and lifelong period groups compared with the control group (all p < 0.05, respectively; Figure 2(c)).

Hepatic lipid drop after DEHP exposure in male offspring (PND 70). (a) Liver sections stained with hematoxylin and eosin (magnification, Χ200; triangle: macro lipid droplets, arrow: micro lipid droplets). The number of (b) macro and (c) micro lipid drop in the liver. Data represent the mean ± standard error (n = 8/group). *p < 0.05, **p < 0.01 for early life- and lifelong period group vs control group.
TG related genes and protein expression in the liver
Genes expression
The early life stage group showed significant overexpression of mRNA encoding 1-acylglycerol-3-phosphate acyltransferase (AGPAT1) and DGAT1 when compared with the control group (all p < 0.05; Figure 3(a)). In the lifelong period group, mRNA expression of acyl-CoA synthetase long chain family member 1 (ACSL1), peroxisome proliferator-activated receptor alpha (PPARα), patatin-like phospholipase domain-containing 2 (PNPLA2), monoacylglycerol acyltransferase 1 (MGAT1), and DGAT1 was significantly increased with that in the control group (all p < 0.05, respectively; Figure 3(a)). DGAT1 mRNA expression was significantly increased compared with that in the early life stage group (p < 0.05; Figure 3(a)). However, SREBP-1c and glycerol-3-phosphate acyltransferase (GPAT) mRNA expression levels were significantly lower than the early life stage group (all p < 0.05; Figure 3(a)).

Effects of DEHP on TG related genes in liver of male offspring (PND 70). (a) The levels of mRNA expression on each gene is normalized to the average of GAPDH, RPLP0, and β-actin in liver (n = 8/group). Data are presented as fold changes from the controls. Protein levels of (b) DGAT1 and (c) SREBP-1c in liver is analyzed by western blotting. Densitometry quantification of DGAT1 protein expression levels normalized to GAPDH. Data represent the mean ± standard error (n = 8/group). *p < 0.05, ** p < 0.01 for DEHP treated groups vs control group; #p < 0.05 for early life stage- vs lifelong period group.
Protein expression
DGAT1 protein expression in early life stage and lifelong period groups was also shown to be significantly higher than the control group (p < 0.05 and p < 0.01, respectively; Figure 3(b)). Protein expression of SREBP-1c in the life long period group showed significantly decreased compared to the control group (p < 0.05, Figure 3(c)).
Discussion
The current study demonstrates that early life stage exposure to DEHP is associated with lipid accumulation in the liver of adult male offspring. It was revealed the increase of micro lipid droplets, TG levels, DGAT1 mRNA and protein expression and decrease of SREBP-1c mRNA and protein expression in DEHP exposure during early life stage and lifelong period group compared to the control group.
TG is the major energy reservoir deposited in the liver, and it is a key component of NALFD. 21 TG synthesis is realized through re-esterification of partial glycerides, which come from partial hydrolysis of pre-existing TGs, and through de novo glyceride synthesis. Lipid droplets, of which TGs are major components, can accumulate when the rates of hepatic fatty acid uptake from plasma or de novo fatty acid synthesis is greater than the rate of fatty acid oxidation or export of hepatic TGs via very low density lipoproteins. In this study, early life stage and lifelong period groups showed significantly increased hepatic TG levels. In line with our study, it has been reported that hepatic TG levels were markedly increased after continuous exposure of adult male rats to DEHP at 5 and 500 mg/kg/day over the course of 8 weeks, 7 and after treatment of HepG2 cells with DEHP 100 μM. 8 It seems that DEHP could induce lipid accumulation in the liver.
Hepatic TG accumulation observed in early life stage and the lifelong period groups might be induced by the detected up-regulation of DGAT1 in the liver. DGAT1 and DGAT2 are mainly located in the endoplasmic reticulum. Those are considered important modulators of energy metabolism because of their functions in catalyzing esterification reactions of fatty acids by utilizing diacylglycerol for TG synthesis. 22 However, DGAT1 cannot compensate for DGAT2 deficiency in vivo. DGAT2-deficient mice showed at least 90% of reduction of TGs in the liver, and died shortly after birth. 16 As the inhibition of DGAT2 directly blocks TG synthesis, suppresses de novo lipogenesis, and stimulates oxidation of fatty acids in hepatocytes,23,24 DGAT2, but not DGAT1, is considered the dominant DGAT enzyme controlling TG homeostasis. 16 However, other studies reported that DGAT1 has a major role in the development of hepatic steatosis through esterification of exogenous, but not endogenous fatty acids.25,26 DGAT1 null mice showed resistance to diet-induced obesity, and decreased hepatic TG levels. 27 In accordance with our data, hepatic TG contents were increased as DGAT1 was overexpressed in mouse 28 and rat hepatoma cells. 29 According to the results presented, overexpression of DGAT1 in early life stage and the lifelong period groups might be related to the observed hepatic lipid accumulation as a consequences of DEHP exposure.
Even if DGAT1 and DGAT2 are functionally linked to TG storage, the sizes of the lipid droplets formed under each scenario are not consistent. Overexpression of DGAT1 induced the formation of small lipid droplets (400–800 nm in diameter), whereas large lipid droplets were observed in DGAT2 expressed cells (typically 1–2 μm).16,30 Moreover, vacuoles in the DEHP-treated group fed a high-fat diet were smaller than those of the high-fat diet only group. 7 Liver fibrosis was also detected in response to treatment with DEHP at 5 and 500 mg/kg/day. In our study, the number of small lipid droplets was significantly increased in early life stage and the lifelong period groups compared with the control group. Although inflammation in the liver tissue was not observed, the increase in small lipid droplets might have resulted from the up-regulation of hepatic DGAT1 mRNA expression.
PPARα is a member of the nuclear receptor superfamily, and has an important role in the lipid metabolism of the liver. It could regulate the activation of ACSL1 in the liver,31,32 and involve the expression of PNPLA2 which is a key enzyme in lipid catabolism in the liver. 33 Activation of PPARα in turn activates fatty acid oxidation through down-regulation of SREBP-1c.34,35 It is considered that PPARα exerts a protective effect against NAFLD. However, the increase of PPARα after DEHP exposure promoted the production of reactive oxygen species and resulted in an increase of hepatic lipids.7,8 SREBP-1c is a key enzyme in lipogenesis that is controlled by insulin and glucose levels. 36 That is, high carbohydrate feeding and fasting leads to elevated and reduced expression of SREBP-1c in animals, respectively. Previous studies suggested that the increase in SREBP-1c and PPARα levels in the liver may account for hepatic lipid accumulation upon DEHP exposure.7,8 However, our data showed that the early life stage group had similar SREBP-1c and PPARα mRNA expression compared with the control group. The lifelong period group showed significantly decreased SREBP-1c mRNA expression and significantly increased PPARα mRNA expression compared with the control. The protein expression of SREBP-1c in the lifelong period group showed significantly down-regulated compared to the control group. The previous study also showed that mono-2-ethylhexyl phthalate, the major metabolite of DEHP, increased TG levels in the HepG2 cells but the protein expression of SREBP-1c was not changed. 37 Thus, SREBP-1c is known as an important gene in lipogenesis, but it may not be in case of not treated with a nutritious diet together.
In addition, exposure to DEHP during an early life stage showed a similar trend with regard to body weight gain. The lifelong period group showed a decrease in body weight gain after the weaning period compared with the control group. The observed decrease in body weight gain might be related to the increase in PPARα mRNA expression and the decrease in SREBP-1c mRNA levels that we observed. A decrease in body weight in male and female offspring was previously reported for perinatal DEHP exposure (1.25 and 6.25 mg/kg/day). 38 However, perinatal exposure to DEHP (300 mg/kg/day from GD 6 to PND 21) did not affect body weight change in another study. 39 Although these studies did not report on hepatic enzyme expression profiles, an activation of PPARα could have induced weight reduction by suppressing de novo fatty acid synthesis.
Importantly, in our study, we identified the key enzyme (DGAT1) responsible for increased synthesis and storage of TGs in the liver after DEHP exposure. Until recently, DGAT2 is a target agent for inhibition of hyperlipidemia because it catalyzes a rate limiting step in the synthesis of TGs. 40 However, an inhibitor of DGAT1 caused a decrease in weight gain and hepatic TGs in mice fed a high-fat diet. 41 In line with our results, inhibition of DGAT1 might prove useful for treatment of hepatic steatosis through a decrease in TG metabolism. In addition, we examined hepatic lipid accumulation in adult male rats after exposure to DEHP during an early life stage as well as over lifelong period. Moreover, previous studies used treatment with DEHP on adult male rats 7 and adult male zebrafish 9 to examine the relationship between DEHP exposure and the development of NAFLD. To our knowledge, this is the first study to investigate the association between exposure to DEHP during a critical period of development and subsequent interference with lipid metabolism in adulthood.
In conclusion, early life exposure to DEHP increases hepatic lipid accumulation in adult male rats, which is presumably mediated by the actions of the DGAT1 enzyme (Figure 4). Although further studies are required to understand the mechanisms of actions of DEHP in early life, DGAT1 may emerge as a new target for treatment of hepatic steatosis induced by environmental chemicals.

The possible mechanism of DEHP on the triglycerides synthesis and lipid droplet accumulation in liver. DEHP: di (2-ethylhexyl) phthalate; DGAT1: diacylglycerol acyltransferase 1; DGAT2: diacylglycerol acyltransferase 2; PNPLA2: patatin-like phospholipase domain-containing 2.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.