
Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive lung disease with high morbidity and mortality. miR-182-5p is overexpressed in several fibrosis-related diseases but its effect in pulmonary fibrosis has not been reported yet. To investigate the function of miR-182-5p in pulmonary fibrosis, we established bleomycin (BLM)-induced fibrotic mice model and transforming growth factor-β1 (TGF-β1)-treated human embryonic lung fibroblasts model. In this study, miR-182-5p was highly expressed in pulmonary tissues of BLM-induced fibrotic mice. The content of hydroxyproline and TGF-β1 was decreased by downregulating the expression of miR-182-5p, indicating that fibrosis was alleviated in mice treated with Lentivirus-anti-miR-182-5p.Quantification of fibrosis-related proteins demonstrated that downregulation of miR-182-5p inhibited the expression of profibrotic proteins (fibronectin, α-smooth muscle actin, p-Smad2/p-Smad3) as well as enhanced the level of Smad7. In vitro assays validated that miR-182-5p was induced by TGF-β1 with the function of promoting fibrosis. In dual-luciferase reporter assay, Smad7 was demonstrated to be negatively regulated by miR-182-5p. Moreover, the effect of knocking down miR-182-5p on inhibiting fibrosis was achieved by upregulating the expression of Smad7. Therefore, miR-182-5p can be regarded as a biomarker of IPF and its inhibition may be a promising therapeutic approach in treating IPF.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and fibrotic lung disease, which is the most common type of idiopathic interstitial pneumonia. 1,2 It is clinically featured by progressive dyspnea and deterioration of pulmonary function. The excessive deposition of extracellular matrix and destruction of alveolar structure lead to decreased lung compliance and gas exchange impairment, eventually resulting in respiratory failure and death of the patients. The morbidity and mortality of IPF increase with age, and it mostly happens among middle-aged and elderly adults (55–75 years old). 3 Patients with IPF have a short median survival time of 3–5 years. 4,5 The risk factors associated with IPF include old age, genetic alterations, smoking history, occupational exposure, and viral infection. 6,7 Several therapies has been applied in treating IPF, such as anti-inflammatory agents (corticosteroids, azathioprine), anti-fibrotic drugs (colchicine, pirfenidone), and immune modulators (nintedanib, interferon gamma-1b). 2 However, the pathogenesis of IPF is still unclear and there are no effective treatments for IPF so far.
MicroRNAs (miRNAs) are a class of single-stranded noncoding small RNAs with the length of 20–23 nucleotides. 8 Through recognizing and binding with the 3′untranslation regions (UTRs) of target mRNAs, miRNAs can meditate the cleavage, degradation, or transcriptional repression of mRNAs. To date, thousands of miRNAs have been found in various species and widely participating in the tissue development process, metabolism process, and regulatory process of multiple diseases. 9,10 It has been reported that the expression of these miRNA molecules is aberrant in diseases like cancer, cardiovascular diseases, and nervous system diseases. 11,12 Besides, the abnormal expression of various miRNAs also exists in IPF. 13 miRNAs such as LET-7d, miR-21, and miR-155 have been demonstrated to be significantly downregulated or upregulated in IPF. 14 Moreover, other possible miRNAs related to IPF are also under study. miR-182-5p was found to have significant upregulated expression in hepatic fibrosis 15 –17 and peritoneal fibrosis. 18 Huang et al. found that overexpressed miR-182 can regulate FOXO1 negatively and prompt the development of hepatic fibrosis. 15 Liu et al. found that miR-182 might play an important role in the process of peritoneal fibrosis. 18 However, the potential effect of miR-182-5p on pulmonary fibrosis has not been reported yet. Although the molecule mechanism of IPF is still dimness, it is certain that transforming growth factor-β (TGF-β) plays an important regulative role in the disease. TGF-β has multiple functions in regulating cell growth and differentiation. 19 Three isoforms have been identified in the TGF-β superfamily: TGF-β1, TGF-β2, and TGF-β3. Among them, TGF-β1 plays a dominant role in promoting fibrotic disorders. It is a key fibroblast-activating factor and has a strong profibrotic effect. In pulmonary fibrosis, the overexpression of TGF-β can increase the synthesis of extracellular matrix or regulate the expression of integrin to accelerate the fibrotic process. 20,21 TGF-β mediates the phosphorylation of Smad2 and Smad3. The phosphorylated Smad2/Smad3 further forms complexes with Smad4 and translocates into the cells to regulate target genes related to fibrosis. By contrast, Smad7 is an important negative feedback regulator in the TGF-β signaling pathway, which can prevent the phosphorylation of Smad2/Smad3 and suppress the TGF-β signaling pathway. 22 Furthermore, Smad7 can inhibit the expression of α-actin activated by TGF-β. miRNAs are known to be critical regulators in the fibrosis process and they can mediate the expression of downstream target genes involved in fibroblast activation, matrix formation, and accumulation. According to reports, Smad7 is one of the downstream target genes of miR-182-5p and their targeting regulatory relationship has been demonstrated in tumor cells. 23 Chung et al. found that Smad7 can reduce renal fibrosis by inhibiting the activation of TGF-β/Smad pathway and affecting the level of related miRNAs. 24 In this study, the function of miR-182-5p in mediating the process of pulmonary fibrosis and the relationship between miR-182-5p and Smad7 were investigated in models of bleomycin (BLM)-induced mice and TGF-β1-treated human embryonic lung fibroblasts (HELFs).
Materials and methods
Establishment of BLM-induced mice models
Six- to-eight-week-old female mice (BALB/c) weighing 20–25 g were randomly divided into four groups (control, BLM, BLM+Lentivirus-negative control (LV-NC), and BLM+LV-anti-miR-182-5p). In each group, six mice participated in the experiment. LV-mediated anti-miR-182-5p interference vector was constructed and packaged. On the first day, mice of the BLM, BLM+LV-NC, and BLM+LV-anti-miR-182-5p groups were intratracheally injected with 50 μL 5 mg/kg BLM (Aladdin, B107423, China) to induce pulmonary fibrosis, while the control group mice were injected with the same volume of normal saline. The next day, the BLM+LV-NC and BLM+LV-anti-miR-182-5p groups were injected with 50 μL 1 × 109 TU/mL LV-NC or LV-anti-miR-182-5p. The control and BLM groups were injected with the same volume of normal saline. Mice of the four groups were executed 21 days later. Their left lungs and serum were collected to investigate the relevant indexes. The animal study was reviewed and approved by the Ethics Committee of Shengjing Hospital of China Medical University.
Determination for the content of hydroxyproline and TGF-β1
For determination of hydroxyproline (HYP), 50 mg tissue from the left lung was hydrolyzed and boiled for 20 min. Then, the pH of the obtained hydrolysate was adjusted to 6.5 according to the manufacturer’s instruction of the HYP Test Kit (Jiancheng, A030-2, China). Activated carbon (60 mg) was added to the hydrolysate and absorbance of the supernatant was measured by ultraviolet (UV) spectrophotometer (Yoke, UV752 N, China) at 550 nm. For determination of TGF-β1, the serum was collected, agglutinated for 30 min at room temperature, and then placed at 4°C overnight. After centrifuging for 10 min at 1000 g, the supernatant serum was collected. Then, 40 μL sample was added to 20 μL 1 N hydrochloric acid (HCl) solution and incubated for 10 min, followed by the addition of 20 μL 1 N sodium hydroxide (NaOH) solution. After activation, 20 μL sample was diluted by adding 480 μL buffer solution and tested by Elisa Kit (Liankebio, EK981, China) according to the instruction.
Histological staining of H&E and Masson’s trichrome
Pulmonary tissues collected from the left lungs of the control, BLM, BLM+LV-NC, and BLM+LV-anti-miR-182-5p groups were fixed by neutral formaldehyde, dehydrated and embedded in paraffin. Tissues in paraffin were cut into slices of 5 µm using a slicer (Leica, RM2235, Germany), then removed to the slides and made into samples of histological staining. Hematoxylin and eosin (H&E) and Masson’s trichrome staining were performed on the tissue slices to indicate the degree of pulmonary fibrosis in the four groups. All samples were observed by an optical microscope (OLUMPUS, BX53, Japan).
Construction of cell models and transfection experiments
HELFs were purchased from CHI SIENTIFIC and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, Logan, UT, USA) containing 10% fetal bovine serum (Sigma, St Louis, MO, USA) at 37°C. The atmosphere of the cell incubator was set as 95% air + 5% CO2. After growing to a suitable density, the cells were digested, dispersed, and inoculated into 6-well plates. In order to establish the corresponding cell model, HELFs were treated with 10 ng/mL TGF-β1 (USCN Life Science, RPA124Hu01, China) for 24 h and the normal HELFs were set as the control group. Transfection experiments were implemented on HELFs in 6-well plates. The cells were cultured in basal medium of DMEM for 1 h before transfection. Then, the mixture of 200 μL Opti-MEM (Gibco, 31985-070, Carlsbad, CA, USA), 200 μL Lipofectamine 2000 (Invitrogen, 11668-019, Carlsbad, CA, USA), and 50 pmol transgenes (NC inhibitor, miR-182-5p inhibitor, NC mimic, miR-182-5p mimic, or Smad7 siRNAs) were added into the plates with continuous gentle shaking. HELFs were further cultured in the incubator at 37°C for 24 h and the corresponding groups were treated with 10 ng/mL TGF-β1 for another 24 h.
Immunofluorescence staining of α-SMA
HELFs slides of control, TGF-β1, TGF-β1+NC inhibitor, and TGF-β1+miR-182-5p inhibitor groups were collected, fixed in 4% paraformaldehyde, and washed in phosphate-buffered saline (PBS) for 3 times. The cell slides were covered by 0.1% Triton X-100 solution, incubated for 30 min, and washed with PBS for 3 times to remove the residual Triton X-100. Then, the cell slides were accordingly incubated with goat serum (Solarbio, SL038, China) for 15 min, 1:300 diluted primary antibody (Bioss, Bs-10196 R, China) overnight at 4°C, and 1:200 diluted fluorescent secondary antibody (Beyotime, A0516, China) for 60 min. 4’,6-Diamidino-2-phenylindole (Beyotime, C1002, China) was used to stain the nuclei of HELFs. Finally, the HELFs slides were treated by antifade mounting medium (C0296-3, Solarbio, China) and observed by a Fluorescence Microscope (BX53, OLUMPUS, Japan).
Real-time PCR
Total miRNA or mRNA was extracted and measured by an UV spectrophotometer (NANO 2000 Thermo Fisher Scientific, Waltham, Massachusetts, USA) to determine the concentration. Reverse transcription from RNA to cDNA was conducted by polymerase chain reaction (PCR; Exicycler 96, BIONEER, Korea) according to the manufacturer’s instruction. Then real-time PCR was implemented by adding with cDNA template, primer (designed by us), SYBR green (S9430; Sigma, St Louis, MO, USA), and Taq PCR MasterMix (PR1702, BioTeke, China). The data were analyzed by 2− ΔΔCt method. The sequences of primers are listed in Table 1.
The sequences of primers.

Western blot
The relative protein contents of lung tissues or HELFs were determined by Western blot. Briefly, Total protein was extracted by incubating the samples with cell lysis buffer (Beyotime, P0013, China) added with 1 mM phenylmethanesulfonyl fluoride for 5 min on the ice. After that, samples were centrifuged in the refrigerated centrifuge (Cence, H-2050 R, China) for 5 min and the supernatant was obtained as the protein extract. Concentration of the proteins was quantitated by BCA Kit (Beyotime, P0011, China); 20–40 μg equal amounts of proteins were diluted with sample loading buffer (Beyotime, P0015, China) and PBS. Then, the sample solutions were treated with 8% or 11% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, IPVH00010, Billerica, MA, USA). After blocking with 3% bovine serum albumin solution (Biosharp, BS043, China) for 1 h, the PVDF membranes were initially incubated with primary antibody and then incubated with secondary antibody. The PVDF membranes were sprinkled with enhanced chemiluminescence (ECL) luminol reagent (Beyotime, P0018, China), exposed in the darkroom, scanned, and analyzed with Gel-Pro-Analyzer to calculate the optical density of target strips. Information about the antibodies is listed in Table 2.
The information of antibodies.
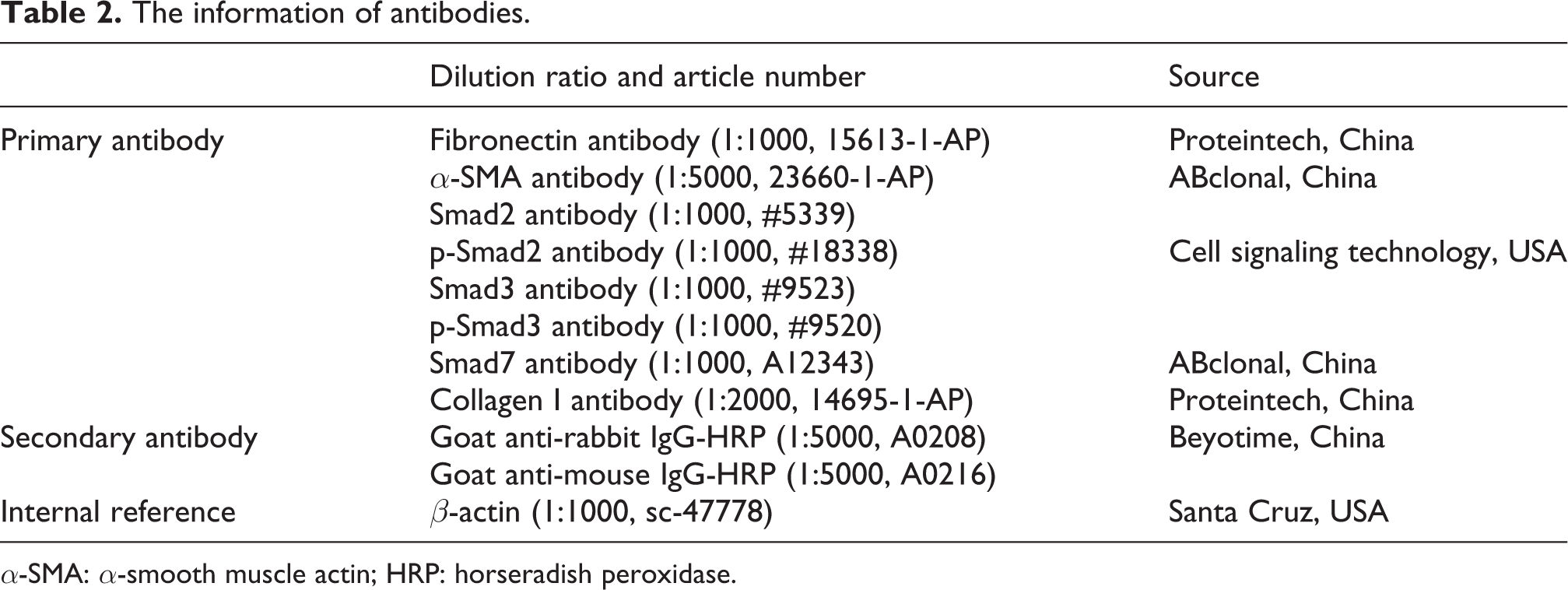
α-SMA: α-smooth muscle actin; HRP: horseradish peroxidase.
Dual-luciferase reporter assay
The targeting relationship between Smad7 and miR-182-5p was examined by dual-luciferase reporter assay. Gene fragment of wildtype Smad7 3′UTR or mutant Smad7 3′UTR was introduced into the luciferase reporter plasmid. HELFs were inoculated into 12-well plate and incubated in serum-free DMEM for 1 h. Then, HELFs were co-transfected with luciferase reporter plasmid and miR-182-5p mimic (or NC mimic). After 48 h, the cells were collected and lysed to measure the luciferase activity by Firefly Luciferase Assay Kit according to the manufacturer’s protocol (E1910, Promega, Fitchburg, WI, USA) and the absorbance was determined by the Microplate Reader (TECAN, M200Pro, Switzerland).
Statistical analysis
Statistical analysis was performed by GraphPad Prism. One-way ANOVA was used to analyze the significance among three or more groups and Student’s t-test was used to analyze the significant between the two groups. Data were represented as mean ± SD and p < 0.05 was considered to have a significant difference.
Results
Downregulation of miR-182-5p suppressed the procession of pulmonary fibrosis in mice
Compared with the control group, the expression of miR-182-5p in pulmonary tissues of BLM-induced fibrotic mice was significantly higher, which verified that the expression of miR-182-5p was upregulated in pulmonary fibrosis. To downregulate the expression of miR-182-5p, fibrotic mice were treated with LV-anti-miR-182-5p. It was found that the expression of miR-182-5pin BLM+LV-anti-miR-182-5p group was greatly decreased, which showed that the injected LV-anti-miR-182-5p worked in mice to downregulate the expression of miR-182-5p while LV-NC did not affect its expression (Figure 1(a)). The content of HYP was also determined in the four groups to indicate the degree of pulmonary fibrosis since HYP was one of the main compositions of collagen (Figure 1(b)). 25 The results showed that the BLM group had a higher content of HYP than normal control, indicating that BLM had induced pulmonary fibrosis in mice. However, after treating with LV-anti-miR-182-5p, the content of HYP was obviously declined, implying the reduction of fibrosis in BLM+LV-anti-miR-182-5p group. According to previous study, TGF-β1 in the TGF-β superfamily is a key profibrotic cytokine in the process of fibrosis. 26 We determined the content of TGF-β1 in the four groups and found that the concentration of TGF-β1 was increased in the BLM group. Besides, the concentration of TGF-β1 went down after treatment with LV-anti-miR-182-5p (Figure 1(c)), which suggests that TGF-β1 was positively correlated with miR-182-5p in fibrotic mice. H&E and Masson’s trichrome staining were performed on the pulmonary tissues. The representative images showed clear pulmonary interstitial thickening and decreased alveoli in the BLM and BLM+LV-NC groups as well as massive aggregation and infiltration of inflammatory cells, which dramatically reduced in the LV-anti-miR-182-5p-treated group (Figure 1(d)).
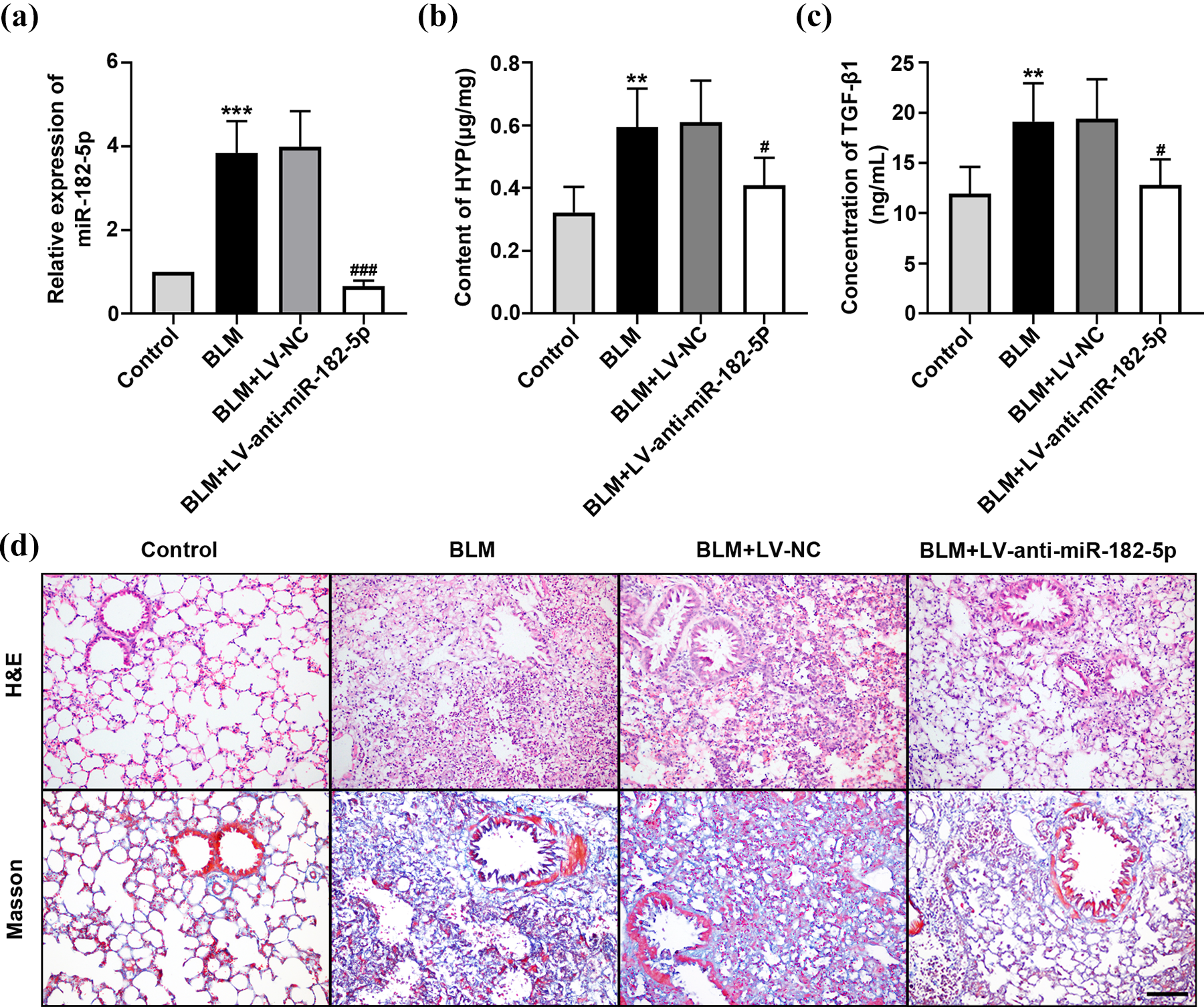
The relative expression of miR-182-5p and pathological changes in pulmonary tissues of normal and BLM-induced fibrotic mice. The mice were injected with BLM, BLM + LV-NC, or BLM + LV-anti-miR-182-5p. The normal mice were set as the control group: (a) the relative expression of miR-182-5p in lung tissues from the mice; (b) the content of HYP in lung tissues of the mice determined by HYP Test Kit; (c) the content of TGF-β1 in lung tissues of the mice determined by ELISA Kit; and (d) H&E and Masson’s trichrome staining of lung tissues from the four groups. Scale bar = 100 μm. *p < 0.05; **p < 0.01; and ***p < 0.001: compared with the control group. #p < 0.05; ##p < 0.01; and ###p < 0.001: compared with the BLM+LV-NC group. BLM: bleomycin; LV-NC: Lentivirus-negative control; LV-anti-miR-182-5p: Lentivirus-anti-miR-182-5p; HYP: hydroxyproline; TGF-β1: transforming growth factor-β1; ELISA: enzyme-linked immunosorbent assay; H&E: hematoxylin and eosin.
miR-182-5p regulated the expression of fibrosis-related proteins in mice
Furthermore, we determined the relative protein expression of fibronectin, α-smooth muscle actin (α-SMA), Smad2, Smad3, p-Smad2, p-Smad3, and Smad7 in pulmonary tissues by Western blot. The expression of fibronectin and α-SMA was upregulated in the BLM and BLM-LV-NC groups. After treating with LV-anti-miR-182-5p, the expression of these profibrotic proteins was significantly downregulated (Figure 2(b) and (c)), which meant the degree of fibrosis was reduced in the BLM+LV-anti-miR-182-5p group. Phosphorylation of Smad2 and Smad3 was an important process in TGF-β signaling pathway of promoting tissue fibrosis. 27 We examined the expression of p-Smad2/p-Smad3 and found that their expression was increased in the BLM and BLM+LV-NC groups (Figure 2(d) and (f)), in which the degree of fibrosis was very high. The BLM+LV-anti-miR-182-5p group exhibited a decreased expression of p-Smad2/p-Smad3 compared to the BLM+LV-NC group. The expression levels of Smad2/Smad3 were comparable between the four groups (Figure 2(e) and (g)). As an important negative feedback regulator, the expression of Smad7 can suppress fibrotic process. 28 It was found that the BLM and BLM+LV-NC groups showed declined expression of Smad7, which in contrast was upregulated in the LV-anti-miR-182-5p-treated group (Figure 2(h)).

Fibrosis-related protein expression in pulmonary tissues of normal and bleomycin-induced fibrotic mice: (a) the protein bands and the relative expression of (b) fibronectin; (c) α-SMA; (d) p-Smad2; (e) Smad2; (f) p-Smad3; (g) Smad3; and (h) Smad7. *p < 0.05; **p < 0.01; and ***p < 0.001: compared with the control group. #p < 0.05; ##p < 0.01; and ###p < 0.001: compared with the BLM + LC-NC group. α-SMA: α-smooth muscle actin; BLM: bleomycin; LV-NC: Lentivirus-negative control.
Downregulation of miR-182-5p inhibited fibrosis in vitro
The effects of miR-182-5p on regulating fibrosis-related proteins (α-SMA, collagen I, fibronectin, Smad2, Smad3, p-Smad2, p-Smad3, and Smad7) in HELFs were further explored. HELFs were treated with TGF-β1, TGF-β1+NC inhibitor, or TGF-β1+ miR-182-5p inhibitor. The images of immunofluorescence staining for α-SMA showed increased fluorescence intensity in the TGF-β1 and TGF-β1+NC inhibitor groups, which indicated that the expression of α-SMA in these two groups was relatively high. But the fluorescence intensity was decreased in the TGF-β1+miR-182-5p inhibitor group (Figure 3(a)). The result of real-time PCR showed that HELFs treated with TGF-β1 had a higher expression of miR-182-5p compared to normal HELFs, proving that TGF-β1 upregulated the expression of miR-182-5p. After treated with miR-182-5p inhibitor, the expression of miR-182-5p was markedly downregulated while NC inhibitor did not impact the expression of miR-182-5p (Figure 3(b)). Western blot analysis informed that the fibrosis-related protein expression of α-SMA, collagen I, fibronectin, p-Smad2, and p-Smad3 was increased by stimulating withTGF-β1, whereas treating with miR-182-5p inhibitor downregulated these indicators (Figure 3(c), (d), (e), (g), (h), (j), and (k)). The expression of Smad2/Smad3 had no significant difference between these groups (Figure 3(g), (i), (j), and (l)). We also found that the expression of Smad7 was significantly decreased in the TGF-β1 group, indicating that TGF-β1 had an inhibiting effect on the expression of Smad7. However, the addition of miR-182-5p inhibitor promoted the expression of Smad7 (Figure 3(f)).

The relative expression of miR-182-5p and fibrosis-related proteins in HELFs. HELFs were treated with TGF-β1, TGF-β1+NC inhibitor, or TGF-β1+miR-182-5p inhibitor. The normal HELFs were set as the control group: (a) representative images of immunofluorescence staining for α-SMA (α-SMA-red, nucleus-blue) in HELFs. Scale bar = 50 μm; (b) the relative expression of miR-182-5pin HELFs. Western blot and semiquantitative analysis of fibrosis-related proteins including (c) α-SMA; (d) collagen I; (e) fibronectin; and (f) Smad7 in HELFs; (g) protein bands of p-Smad2 and Smad2. The relative expression of (h) p-Smad2 and (i) Smad2 in HELFs; (j) protein bands of p-Smad3 and Smad3. The relative expression of (k) p-Smad3 and (l) Smad3 in HELFs. *p < 0.05; **p < 0.01; and ***p < 0.001: compared with the control group. #p < 0.05; ##p < 0.01; and ###p < 0.001: compared with the TGF-β1 group. HELF: human embryonic lung fibroblast; TGF-β1: transforming growth factor-β1; TGF-β1+NC: transforming growth factor-β1+negative control; α-SMA: α-smooth muscle actin.
Smad7 was a direct target of miR-182-5p
The predicted binding site of wildtype Smad7 3′UTR or mutant Smad7 3′UTR with miR-182-5p is shown in Figure 4(a). The relative luciferase activity of Smad7 3′UTR(WT)+miR-182-5p mimic group was significantly lower than Smad7 3′UTR(Mut)+miR-182-5p mimic group. Besides, co-transfecting with Smad7 3′UTR(WT)+NC mimic or Smad7 3′UTR(Mut)+NC mimic did not affect the luciferase activity (Figure 4(b)). These results indicated that Smad7 was directly regulated by miR-182-5p and the predicted fragment was exactly their binding site. In order to make further demonstration, we transfected HELFs with NC mimic, miR-182-5p mimic, NC inhibitor, or miR-182-5p inhibitor and quantified the relative expression of Smad7 by real-time PCR and Western blot. It was found that the expression of Smad7 was much lower in NC mimic-treated cells than in miR-182-5p mimic-treated cells. Conversely, the expression of Smad7 was greatly increased in miR-182-5p inhibitor-treated cells compared to NC inhibitor-treated cells (Figure 4(c) and (d)). Therefore, it was confirmed that the overexpression of miR-182-5p suppressed the expression of Smad7 while the silencing of miR-182-5p enhanced the expression of Smad7. Moreover, it was also revealed that miR-182-5p inhibited the function of Smad7 through mediating the degradation and repressing the transcription of Smad7 mRNA. So Smad7 was a specific target of miR-182-5p and their relationship was negatively correlated.

The targeting relationship between Smad7 and miR-182-5p. HELFs were transfected with NC mimic, miR-182-5p mimic, NC inhibitor, or miR-182-5p inhibitor: (a) predicted binding sites of miR-182-5p with wildtype Smad7 3′-UTR and mutant Smad7 3′-UTR; (b) relative luciferase activity of Smad7 measured by luciferase reporter assay; (c) the relative mRNA expression of Smad7 measured by real-time PCR; and (d) the protein band and relative protein expression of Smad7 measured by Western blot. *p < 0.05; **p < 0.01; and ***p < 0.001: compared with the Smad7 3′-UTR(Mut)+miR-182-5p mimic group. #p < 0.05; ##p < 0.01; and ###p < 0.001: compared with the NC mimic group. &p < 0.05; &&p < 0.01; and &&&p < 0.001: compared with the NC inhibitor group. HELF: human embryonic lung fibroblast; NC: negative control; UTR: untranslation region; PCR: polymerase chain reaction.
Knockdown of miR-182-5pinhibited fibrosis through the regulation of Smad7
In this study, we found that miR-182-5p regulated the expression of Smad7 and profibrotic proteins in mice and HELFs. To clarify their regulatory relationship, we transfected HELFs with different Smad7 siRNAs and selected the Smad7 siRNA with the highest efficiency of knocking down Smad7. The results of real-time PCR and Western bolt showed that the expression of Smad7 was the lowest in cells transfected with Smad7 siRNA-2 (Figure 5(a) and (b)). Then, we treated HELFs with TGF-β1+miR-182-5pinhibitor+NC siRNA or TGF-β1+miR-182-5p inhibitor+Smad7 siRNA and determined the level of relative proteins. The results revealed that HELFs treated with Smad7 siRNA exhibited relatively high expression of profibrotic proteins (fibronectin, α-SMA, and collagen I) and low expression of Smad7, whereas HELFs treated with NC siRNA exhibited lower expression of profibrotic proteins and high expression of Smad7, although they were both treated with TGF-β1 and miR-182-5p inhibitor (Figure 5(c)).These results demonstrated that downregulating the expression of miR-182-5p suppressed the process of fibrosis, and highly expressed Smad7 played an intermediary role in the regulation. The inhibition of fibrosis would not realize without the function of Smad7, even though the expression of miR-182-5p was downregulated, because Smad7 is an important negative feedback regulator in TGF-β signaling pathway to inhibit fibrosis. 29
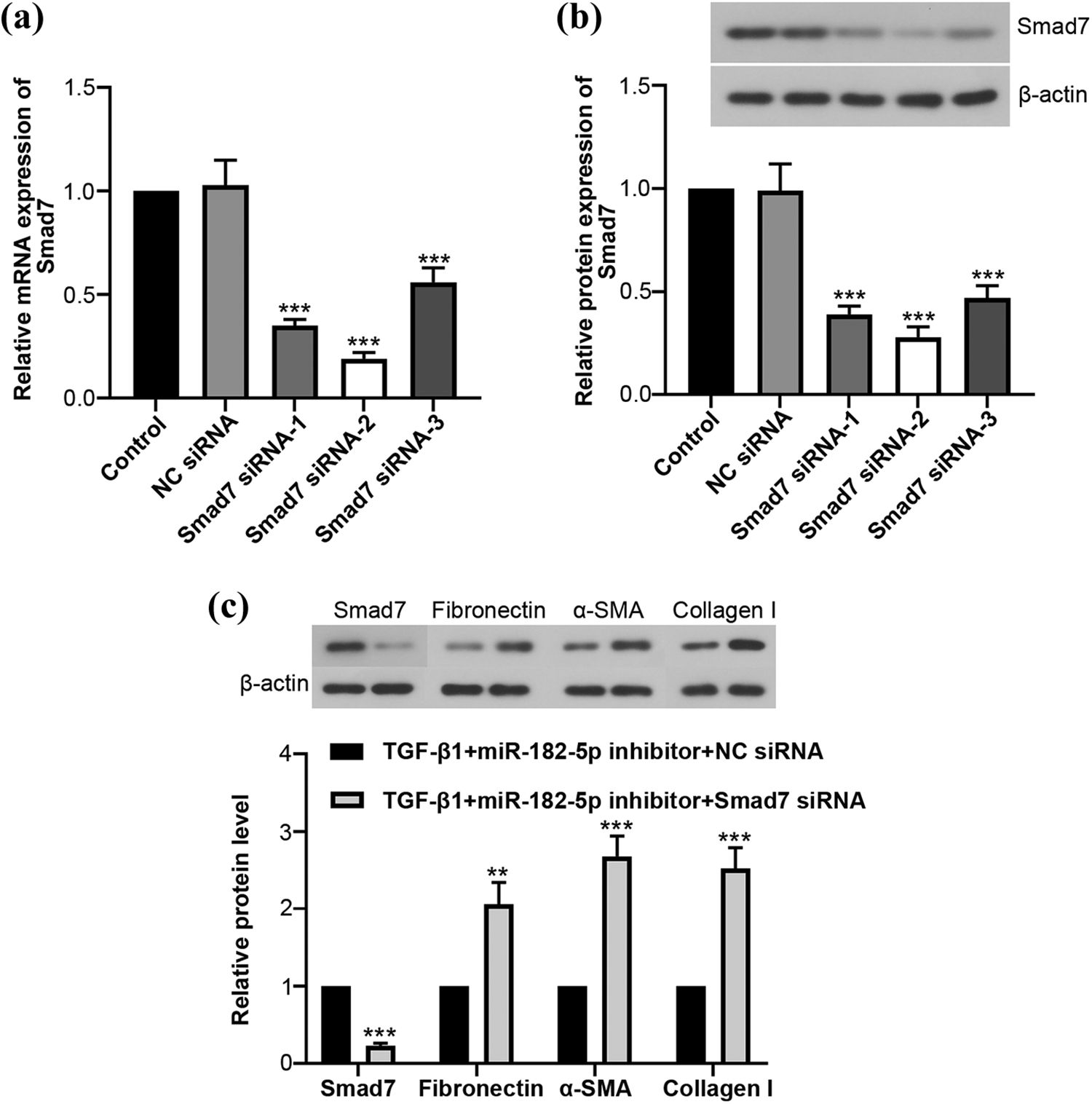
Suppressing the expression of Smad7 led to fibrosis-related protein levels increasing in HELFs. HELFs were treated with TGF-β1+miR-182-5p inhibitor+NC siRNA or TGF-β1+miR-182-5p inhibitor+Smad7 siRNA: (a) the relative mRNA expression of Smad7 in HELFs measured by real-time PCR; (b) the relative protein expression of Smad7 in HELFs measured by Western blot; (c) the relative protein levels of Smad7, fibronectin, α-SMA, and collagen I in HELFs. *p < 0.05; **p < 0.01; and ***p < 0.001: compared with the control group. #p < 0.05; ##p < 0.01; and ###p < 0.001: compared with the TGF-β1+miR-182-5p inhibitor+NC siRNA group. HELF: human embryonic lung fibroblast; TGF-β1: transforming growth factor-β1. mRNA: messenger RNA; PCR: polymerase chain reaction.
Discussion
In this study, we established the BLM-induced mice model of pulmonary fibrosis and TGF-β1-treated HELF cell model to investigate the role of miR-182-5p in mediating the fibrotic process. In mice of model group, the contents of HYP and TGF-β1 were greatly increased, which were important indicators of fibrosis. 25 –30 H&E staining also confirmed the severe fibrosis induced by BLM. These results demonstrated the successful establishment of fibrotic mice model. HELF cell model was also successfully established with the dramatic increase of the profibrotic proteins α-SMA, collagen I, and fibronectin. 31,32 miR-182-5p was found to be dramatically upregulated in BLM-induced mice and TGF-β1-treated HELF cells. Moreover, it was found that inhibition of miR-182-5p could reduce fibrosis through regulating the TGF-β1 signaling pathway.
IPF is a chronic lung disease which results in the deterioration of pulmonary function, respiratory failure, and even death of patients. Anti-inflammatory therapy and anti-fibrotic therapy have been applied to treat patients with IPF. 2 However, significant individual differences and poor prognosis usually occur among patients, which makes it difficult to achieve ideal therapeutic effects. The pathological mechanism of IPF is not completely understood and there are no effective therapies to treat this disease. In order to develop novel approaches to treat IPF, a growing number of researches have focused on studying fibrosis-related growth factors and signaling pathways to find a breakthrough. 26 TGF-β1 is a member of the TGF-β superfamily. With the understanding on the pathogenesis of IPF deepening, TGF-β1 has been found to be a master profibrotic growth factor in the procession of fibrosis, which regulates cell proliferation, differentiation, and apoptosis. 30 The overexpression of TGF-β1 leads to the deposition of collagen and induces multiple fibrotic diseases. 33 In our study, TGF-β1 was normally expressed in pulmonary tissues of healthy mice while it was dramatically increased in BLM-induced fibrotic mice. This finding verified that upregulation of TGF-β1 promoted fibrosis in mice.
TGF-β1 has various functions of regulating fibroblast-type cells on their activation, inducing fibroblasts to generate cytokines, reactive oxygen species, and chemokines. 30 These factors promote the accumulation of extracellular matrix, reduce the degradation of extracellular matrix, stimulate tissues, and lead to fibrosis. In this research, we found that the expression of profibrotic proteins (fibronectin, α-SMA, collagen I, p-Smad2, and p-Smad3) was significantly upregulated in HELFs treated with TGF-β1, demonstrating the profibrotic effect of TGF-β1 in vitro. According to previous reports, TGF-β1 can function through canonicalTGF-β1/Smad signaling pathway or noncanonical Smad-independent pathways. 34 In the TGF-β1/Smad signaling pathway, Smad2 and Smad3 are activated into phosphorylated Smad2/Smad3, which further combine with Smad4 to form a complex, translocate into the nucleus, and mediate target genes. 35 It was found in our study that the levels of p-Smad2/p-Smad3 were remarkably improved in pulmonary fibrotic tissues of mice and HELFs treated with TGF-β1. Besides, the levels of p-Smad2/p-Smad3 were positively correlated with TGF-β1. These results indicated that TGF-β1/Smad signaling pathway was involved in BLM-induced pulmonary fibrosis of mice.
Smad7 has been reported to be a negative regulator in TGF-β1/Smad signaling pathway, which suppresses the phosphorylation of Smad2/Smad3 and blocks TGF-β1 signaling. 29 In this study, the level of Smad7 was downregulated in TGF-β1-treated HELFs and in fibrotic pulmonary tissues of mice, showing that the expression of Smad7 was greatly inhibited by TGF-β1 in pulmonary fibrosis. Since the deficiency of Smad7 aggravated pulmonary fibrosis, more researches had to be conducted to investigate whether the upregulation of Smad7 could conversely suppress fibrosis. miRNAs are key regulators in the process of lung fibrosis, which play important roles in mediating the expression of target genes. 36 It has been demonstrated that the expression of numerous miRNAs is upregulated or downregulated in IPF, including LET-7, miR-21, miR-29, and miR-155. 13 miRNAs can mediate the fibrotic process through inhibiting or promoting the downstream signaling ofTGF-β1. 37,38 As a critical regulator in TGF-β1/Smad signaling pathway, the level of Smad7 can be modulated by miRNAs. 39
In our study, we found that the expression of miR-182-5p was dramatically increased in BLM-induced fibrotic mice accompanied with the reduction of Smad7. To validate whether the overexpression of miR-182-5p promoted fibrosis, the mice were treated with BLM+LV-anti-miR-182-5p and their pulmonary tissues were analyzed to determine profibrotic indicators. It was shown that LV-anti-miR-182-5p inhibited the expression of miR-182-5p as well as increased the level of Smad7, which also reduced the degree of pulmonary fibrosis in mice. We validated the experimental conclusions in HELF models and found that the expression of miR-182-5p was negatively correlated with Smad7. Then, we proved the targeting relationship between Smad7 and miR-182-5p and confirmed that miR-182-5p negatively regulated Smad7 expression by interacting with its 3′UTR. Moreover, we demonstrated that miR-182-5p modulated the levels of profibrotic proteins through the regulation of Smad7, which was an important regulator in TGF-β1/Smad signaling pathway. Approaches aimed at regulating miRNAs to treat fibrosis-related diseases are being widely studied. Li et al. investigated the function of miR-433 in renal fibrosis and found that miR-433 promote fibrosis through TGF-β1/Smad3 signaling pathway. 40 However, the difference in miRNAs regulation between mice and human is unclear. The function of miRNAs is regulated by multiple factors in vivo and more research need to be done on clinical trials.
In conclusion, this research illustrated that the overexpression of miR-182-5p led to deterioration of pulmonary fibrosis through inhibiting the expression of Smad7. Whereas inhibiting the expression of miR-182-5p upregulated the level of Smad7 and reduced pulmonary fibrosis through TGF-β1/Smad signaling pathway. The finding provided a promising biomarker for the diagnosis and prognosis of pulmonary fibrosis. More importantly, the inhibition of miR-182-5p may be a prospective approach to treat IPF or normal pulmonary fibrosis.
Footnotes
Author contributions
MT and YC contributed to the conception of the study. YC and QZ designed the experiment. QZ, YZ, and ZY performed the experiment. YZ and ZY analyzed the data. YC and QZ drafted the manuscript. MT revised the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the National Natural Science Foundation of China (No. 81400042) and the Science and Technology Project of Department of Education, Liaoning Province (No. L2013299).