
Abstract
Introduction:
Tumor microenvironment is known to alter the anticancer drug efficiency. One of the factors that get altered in cancer microenvironment is glucose concentration. Butein, an active principle from plant, known to have anticancer effect against different types of tumor. The objective of the study is to determine the effect of butein on glucose exposed non-small cell lung cancer cells (NSCLCCs).
Methods:
The current study deals with the effect of butein (6.25–50μM) on NSCLCCs treated with different concentrations (0–40 mM) of glucose.
Results and discussion:
Glucose concentration, 0 mM and 40 mM, was found to be lethal at 72 h. Viable cell numbers were statistically increased in 5-mM, 10-mM, and 20-mM glucose-treated cells. Butein at 12.5 µM inhibits (p < 0.05) glucose-induced cell proliferation. Butein inhibits glucose-induced proliferation through DNA damage and oxidative stress. Mitochondrial reactive oxygen species (ROS) level was elevated in 20-mM glucose-treated cells when compared to 5-mM glucose-treated cells, whereas butein treatment further increases glucose-induced mitochondrial ROS. Pharmacological inhibitor of glycolysis, such as 2-deoxy glucose (2-DG), was found to inhibit (p < 0.05) glucose-induced cells proliferation. Furthermore, 2-DG and butein showed synergistic anticancer effect. Butein treatment increases p38 phosphorylation. Inhibition of p38 phosphorylation and antioxidant pretreatment partially revert the glucose-induced cell proliferation. However, inhibition of p38 phosphorylation combined with antioxidant pretreatment completely reverts the anticancer effect of butein. The present study concludes through the evidence that butein could serve as a potential anticancer compound in tumor microenvironment.
Introduction
New therapeutic strategies are essential for non-small cell lung cancer cells (NSCLCCs) because of poor survival of patients, which is less than 5 years. 1 Further, it is noticed that recurrence happens in 30–60% of the patients. 1 –5 The above statements demand the new approach to curtail this cancer. It is known that cancer cells have increased glycolysis; on the other hand, oxidative phosphorylation is reduced in NSCLCCs. 6,7 Cancer cells consequently overexpresses hexokinase (HK) in comparison to the normal cells. In anaerobic glycolysis, isoenzyme HK II is responsible for glycolysis. Further, HK II is responsible for “Warburg” effect. Considering the importance of aerobic glycolysis, different strategies were followed to manage the glycolytic pathway so that cancer cell proliferation could be inhibited. 8,9 For example, miR-143 expression is inversely proportional to HK II expression. 10,11 Another example is the use of miR-122 in liver cells that control aldolase A activity. 12 Different glucose transporter regulators are also targeted through different miR’s to control the cancer cell proliferation. 13 –15 On the whole, promising results were obtained through different mi-RNA to target cancer cell proliferation. However, it will take time to explore the same in human subjects. One of the alternative approaches to control metabolic alteration in cancer cells is through phytochemicals.
Plants such as Semecarpus anacardium, Caragana jubata, and Rhus verniciflua are well-known for their medicinal property, mainly for their anticancer effect. 16 –18 One of the major active constituents present in these plant materials is butein. 16 Butein shows its anticancer effect against wide range of cancers, namely, colon, liver, breast, lung, and so on. 16,19,20 Major mechanism that involves in anticancer effect of butein is through STAT-3 regulation and mitochondria-mediated apoptosis. 21,22 Further, it is reported that butein increases intracellular reactive oxygen species (ROS) in different cancer cells. 23 Studies have shown that butein exposure increases extracellular signal-regulated kinases (ERK) and p38 phosphorylation and increased expression of death receptors such as tumor necrosis factor-related apoptosis-inducing ligand on the cell surface. 16,23 –25 Since increased intracellular ROS is considered to be one of the main mechanisms that involve in anticancer effect of butein, studies show that antioxidants such as N-acetylcysteine (NAC) and reduced glutathione pretreatment reverts the butein-induced cancer cell death. Other studies show that cell cycle arrest, anti-inflammatory effect, and inhibition of angiogenesis are other pathways through which butein could exert its anticancer activity. In nutshell, butein is known to be the potential anticancer drug candidate, and additional investigations are warranted to bring this compound in clinical use.
In the present study, the effect of butein on tumor microenvironment is analyzed. One of the factors that is noticed in solid tumors is gradient in glucose concentration. In order to mimic the in vivo solid tumor, NSCLCCs were exposed to different concentrations of glucose. Effect of butein was assayed in terms of cell viability, cytosolic oxidative stress, DNA damage, mitochondrial membrane potential, different antioxidant enzyme activities, mitochondrial ROS (mROS), and signaling mechanisms.
Materials and methods
A549, a human NSCLCC line, was procured from American-type culture collection (ATCC). Cells were maintained in normal Dulbecco’s modified Eagle’s (DME) medium containing 10% of fetal bovine serum (FBS), 100 U/ml penicillin, and 100 mg/ml streptomycin in a humidified 5% carbon dioxide incubator. At 80% confluency, cells were subcultured using 0.5% trypsin and 0.02% ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, St Louis, Missouri, USA). Cells used in these experiments were between 10 and 20 passages (after obtaining the cells from ATCC). Phosphate-buffered saline (PBS) was used to wash the cells twice before being exposed to different concentrations of glucose. Cells were plated in 48-well (2.5 × 104 cells per well) or 24-well (5.0 × 104 cells per well) cell culture plate or 6 mm (1.0 × 106 cells per plate) dishes and allowed to grow until it reaches approximately 60% confluence, then washed twice with 1× PBS and treated with glucose-free DME medium (Invitrogen, Carlsbad, CA, USA) or DME medium containing different concentrations of glucose, that is, 5 mM, 10 mM, 20 mM, and 40 mM. Butein (Sigma-Aldrich) and/or 2-deoxy glucose (2-DG) (Sigma-Aldrich) were also used in different concentrations to treat the cells. The 500X protease inhibitor cocktail (Biovision, Milpitas, California, USA) and SB203580 (Calbiochem, La Jolla, California, USA) were commercially obtained. P-p38 antibody (anti-rabbit), P-ERK antibody (anti-rabbit), and antibody against β-actin (anti-rabbit) were all procured from Cell Signaling Technology, Inc. (Beverly, Massachusetts, USA)., Peroxidase-linked anti-rabbit immunoglobulin G (IgG) antibody was procured from Sigma-Aldrich Chemical Company.
Cell viability by MTT assay
Viable cells were quantified on the basis of reduction of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) in living cells. 26 Cells were exposed to different (0–40 mM) concentrations of glucose for 0 h, 24 h, 48 h, and 72 h and butein (6.25–50 µM) or 2-DG alone for 48 h. The experimental medium was replaced with 0.5 mg MTT/ml in DME media, and incubation was continued for 4 h at 37°C. MTT-containing medium was removed after 4 h, cells were lyzed, and the intracellular formazan product was solubilized in isopropanol for quantification at optical density at 560 nm (OD560) with background subtraction at OD690. All the assays were performed in triplicate to confirm the results.
LDH activity assay through kit method
A standard kit that is available commercially (Catalog number: C0016; Beyotime Institute of Biotechnology, Nanjing, China) was used to determine the activity of lactate dehydrogenase (LDH).
Immunohistochemical quantification of 8-OHG
The 8-hydroxy-2′-deoxyguanosine (8-OHG) was quantified through established protocols. 27 Cells grown in glass coverslips were incubated with glucose (5 mM, 10 mM, and 20 mM) for 48 h and then treated with butein (12.5 µM, 25 µM, and 50 µM) for 24 h. Following the end of the study, 70% ethanol was used to fix the cells and 0.2% Triton-X in PBS was used for permeabilization. After permeabilization, nonspecific sites were blocked with 10% goat serum; finally, at room temperature, anti-8-oxo-dG (1:1000; GTX41980; Gene Tex, Irvine, California, USA) was applied overnight. After a series of PBS wash (thrice), cells were incubated with goat anti-mouse IgG (A3682; Sigma-Aldrich) conjugated with rhodamine. The nucleus was stained with Hoechst 33456 (Cell Signaling Technology Inc.). To find positive cells and analysis, 10 different fields were selected randomly and examined under the fluorescence microscope at 20× magnification.
DNA damage (strand break) determination by comet assay
DNA strand break was assayed using comet assay, as explained by Suyavaran et al. 28 Cells were treated with different glucose (5 mM, 10 mM, and 20 mM) for 48 h and then treated with butein (12.5 µM, 25 µM, and 50 µM) for 24 h. The experimental cells were collected through trypsin, washed with PBS, and pelleted after centrifugation at 80 × g for 2 min. Around 15,000 cells were mixed with 0.65% low-melting agarose. These cells were employed onto a glass microscope slide that was coated previously with normal melting point agarose (1%). Coverslip was placed on top of these cells, at 4°C for 10 min. Agarose was allowed to set and coverslip was removed. Newly made cell lysing solution (100-mM disodium EDTA, 10-mM Tris–hydrochloride (HCl), 2.5-mM sodium chloride, and 10% dimethyl sulfoxide) was used to lyse the cells, which was done for 3 h. After being rinsed with distilled water and electrophoresis buffer (pH > 13, 300-mM sodium hydroxide, and 1-mM EDTA), the slides were subjected to electrophoresis for 20 min (25 mV, 300 mA). After electrophoresis, 0.4 M Tris-HCl buffer, pH 7.5, was used to neutralize the cells in the slide. After that, the slides were rinsed with distilled water, methanol, and then stained with ethidium bromide.
A CY3-Rhodamine filter cube equipped upright fluorescence microscope was used to score the slides with the help of an image analysis system. The magnification used was 400×. We have determined the tail length by estimating the distance between the nucleus (the head) and the end of the tail. The distance between the left edge of the entire image and the center of the head determines the position of the head. Tail DNA (%) and tail length (migration of the DNA away from the nucleus, µm) were assessed in the images from 100 cells (50 each from duplicate slides).
Mitochondrial membrane potential (JC-1 staining)
The procedure as explained by Preedia Babu et al. 29 was followed to determine and analyze the mitochondrial membrane potential. In detail, cells were exposed to glucose (5 mM, 10 mM, and 20 mM) for 48 h, then treated with butein (12.5 µM, 25 µM, and 50 µM) for 24 h at 37°C, and then treated with JC-1 dye (1 μg/ml) and 1 μg/ml Hoechst 33456 (Cell Signaling Technology, Inc.) for 60 min. Healthy mitochondria was analyzed by a membrane permeable widely using JC-1 dye. Potential-dependent accumulation in mitochondria takes place due to JC-1 dye, designated by an emission shift from green fluorescence (approximately 529 nm) to red fluorescence (approximately 590 nm). Subsequently, depolarization of mitochondrial is signified by a reduction in the intensity ratio of red/green fluorescence. After JC-1 dye staining, fluorescence microscope with blue, green, and red filters was used to observe and analyze the cells.
Intracellular ROS measurement
Cells loaded with 2′,7′-dichlorohydrofluorescein diacetate (DCFH-DA) were subjected for the fluorometrical assay of intracellular ROS presence. 29 In general, cellular esterases hydrolyze the cell permeable DCFH-DA to dihydro-2′,7′dichlorofluorescin, which undergoes oxidation by hydrogen peroxide that produces a fluorescent product, 2′,7′-dichlorofluorescein (DCF). Cells were exposed to glucose (5 mM, 10 mM, and 20 mM) for 48 h and then treated with butein (12.5 µM, 25 µM, and 50 µM) for 24 h, washed with PBS, and then supplied with 10-µmol/L DCFH-DA in DME medium (serum-free) that is devoid of phenol red and contains 0.1% BSA. After 30 min, cells were washed with PBS (thrice) at 37°C. The fluorescence intensity of DCF was evaluated at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. The background fluorescence intensity in the control wells without DCFH-DA was deducted in all cases.
Spectrophotometer assay of antioxidant enzyme activity
The cells were exposed to glucose (5 mM, 10 mM and 20 mM) for 48 h and then treated with butein (12.5 µM, 25 µM, and 50 µM) for 24 h. After the experimental period, cell lysate was prepared. The procedure described by Misra and Fridovich 30 was used to evaluate the superoxide dismutase (SOD) activity. One SOD activity unit is defined as the amount of enzyme that reduces the rate of auto-oxidation of epinephrine by 50%. Previously described assay procedure was followed for catalase (CAT) activity. 31 Glutathione peroxidase (GPx) activity was estimated using previously published protocol. 32 Massey and Williams’ methodology was used to assay the glutathione reductase (GR) activity. 33 The protein content was determined as mentioned in manufacturer’s procedure (Cayman Chemical Company, Ann Arbor, Michigan, USA).
Measurement of mROS
The cells were treated with glucose (5 mM, 10 mM, and 20 mM) for 48 h and then treated with butein (12.5 µM, 25 µM, and 50 µM) for 24 hr. After the experimental period (72 h), cells were collected. The intact mitochondria was isolated through commercially available kit with manufacturer instruction (catalog number: 89874) obtained from ThermoFisher Scientific (Waltham, Massachusetts, USA). Amplex Red Hydrogen Peroxide/Peroxidase assay kit (ThermoFisher Scientific) was utilized to analyze the reverse flow of electrons, ROS generation at complex III and complex I in mitochondria. The mentioned assays were executed at room temperature (37°C).
Western blot analysis for P-p38 and ERK proteins
The cells after being treated with indicated concentration of butein and/or 2-DG were washed with PBS (ice-cold), lysed in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (20 mM, pH 7.5), 10-mM potassium chloride, 1.5-mM magnesium chloride, 1-mM Ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 1-mM EDTA, 1-mM Phenylmethylsulfonyl fluoride (PMSF), 5-μl/ml protease inhibitor cocktail (P-8340; Sigma-Aldrich) and 4-mM DTT by forcing 20 times through a needle (27 gauge), and incubated at 4°C for an hour. Following a 15-min centrifugation at 11,000 × g, the supernatant containing protein (30 μg) was electrophoresed on a 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis. The separated proteins were transferred to Immobilon-P membrane (Millipore, Bedford, Massachusetts, USA). After blocking with 5.0% bovine serum albumin (BSA) in Tris-buffered saline, the membrane was treated with commercially available antibodies raised against P-p38 and P-ERK (all at 1:1000 dilution), and peroxidase-linked secondary antibodies (1:25,000). Enhanced chemiluminescence kit (Amersham-Pharmacia, Piscataway, NJ, USA) and Kodak X-Omat XAR film (Eastman Kodak Co., Rochester, New York, USA) were used for detection. To confirm equal loading, the membranes were probed for the expression of β-actin.
Statistical analysis
Values are expressed in the form of mean ± standard deviation. All the experiments were repeated thrice at least. The statistical analysis of differences between experimental groups was performed by one-way analysis of variance with the Bonferroni multiple comparison posttest, using SPSS 15.0 software for windows (Cary, North Carolina, USA). A p value of less than 0.05 (p < 0.05) was considered statistically significant.
Results
Effect of glucose on NSCLCCs proliferation
Figure 1(a) shows the effect of glucose on NSCLCCs proliferation. Cells were treated with DME (glucose-free) medium that contains 10% FBS and it is considered as 0-mM glucose medium. Cells were treated with media of 0–40-mM glucose concentration for 72 h. The 0-mM and 40-mM glucose-treated cells did not show an increase in cell number at 24 h, 48 h, and 72 h when compared with 0 h. On the other hand, 0-mM and 40-mM glucose treatment for 48 h and 72 h significantly increased the level of LDH activity (Figure 1(b)) when compared with 0 h. These results indicate that very low concentration (0 mM) and very high concentration (40 mM) of glucose are lethal to NSCLCCs. We did not proceed with these lethal doses for our further studies. We have conducted our studies with glucose concentrations of low (5 mM), medium (10 mM), and high (20 mM) treatment for up to 72 h. As seen in Figure 1(a), statistically significant (p < 0.05) level of cell number was observed in 24 h of medium (10 mM) and high (20 mM) concentration of glucose treatment when compared with 0 h. Increasing the duration of the glucose exposure further increases the cell number (p < 0.001). There was no statistically significant change in LDH activity. Figure 2 shows the cell viability (a) and LDH activity (b) in butein alone treated cells. Statistically significant (p < 0.05) level of cell viability was decreased in 50-µM butein-treated cells at 24 h when compared with control cells. When duration was increased, cell viability was reduced (p < 0.05) even at 25-µM butein-treated cells. On the other hand, low concentration of butein was found to be mitogenic (p < 0.05). Similar to cell viability, LDH activity also increased (p < 0.05) in high concentration of butein-treated cells at 24 h. Our present finding correlates with previous literature. 16 Since 25-µM butein is found to be nontoxic at 24 h, for further experiment, we have exposed the cells to glucose for 48 h, then tested with butein for 24 h to find the effect of butein on glucose-induced alterations.
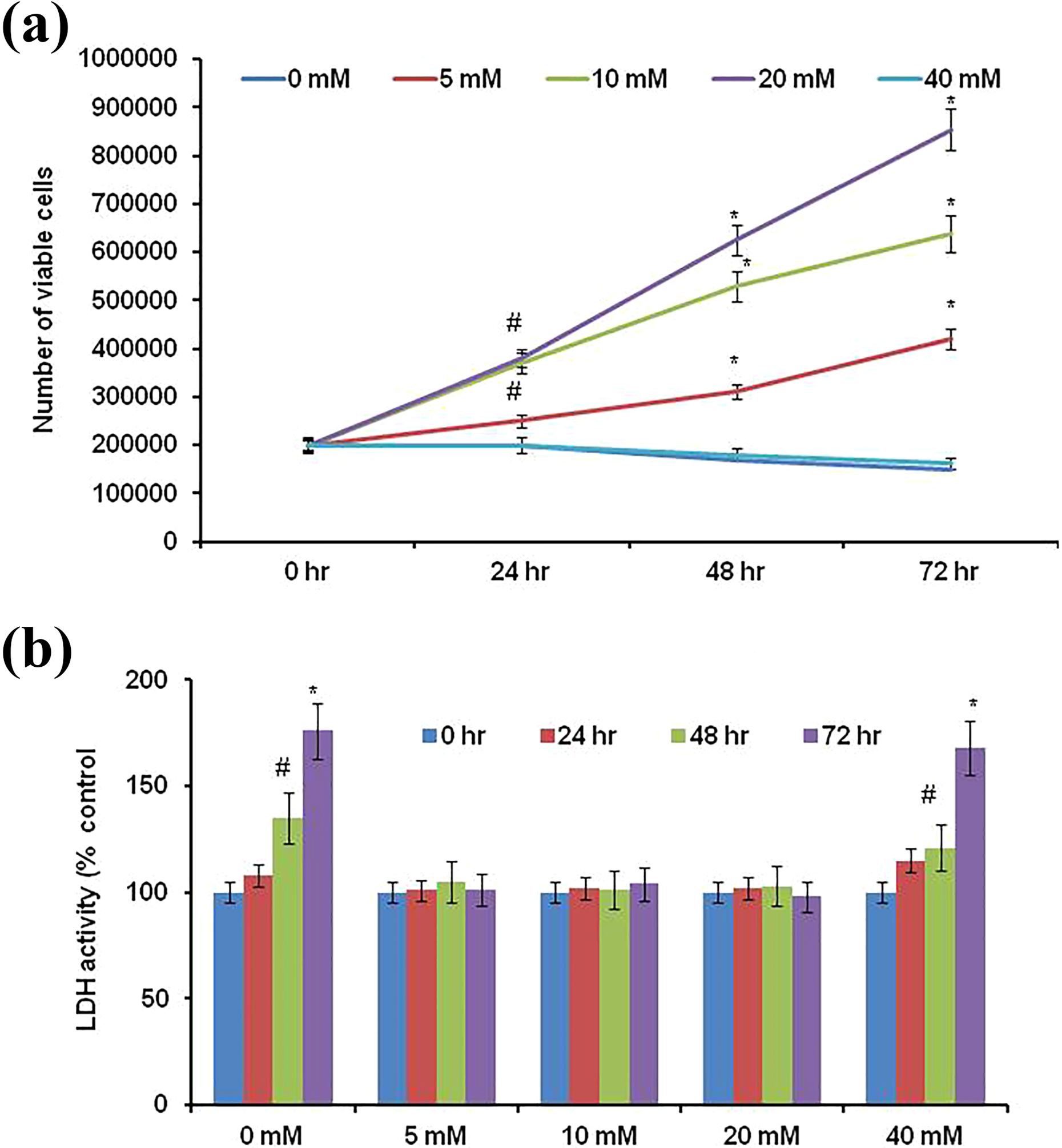
Cell number (a) and LDH activity (a) in different concentrations of glucose-exposed cells. Experiments were repeated four times with triplicate in each time (n = 4). Cells were treated with 0 mM, 5 mM, 10 mM, 20 mM, and 40 mM for 24 h, 48 h, and 72 h. Viable cell number and LDH activity was calculated at 24 h, 48 h, and 72 h. Values are expressed as mean ± SD. Statistical significance was calculated by one-way ANOVA with the Bonferroni multiple comparison posttest. #p < 0.05; *p < 0.001 versus respective 0-h treatment. LDH: lactate dehydrogenase; SD: standard deviation; ANOVA: analysis of variance.

Cell number (a) and LDH activity (b) in different concentrations of butein alone-treated cells. Experiments were repeated four times with triplicate in each time (n = 4). Cells were treated with 0 µM, 6.25 µM, 12.5 µM, 25 µM, and 50 µM for 24 h and 48 hr. Cell viability and LDH activity was calculated at 24 h and 48 h. Values are expressed as mean ± SD. Statistical significance was calculated by one-way ANOVA with the Bonferroni multiple comparison posttest. #p < 0.05; *p < 0.001 versus respective 0-h treatment. LDH: lactate dehydrogenase; SD: standard deviation; ANOVA: analysis of variance.
Glucose-induced oxidative DNA damage and attenuating effect of butein
Glucose-treated cells were exposed to three different concentrations of butein (12.5 µM, 25 µM, and 50 µM) for 24 h. Butein effect was observed in terms of cell viability. Figure 3(a) shows the cell viability of butein-treated cells. Cells treated with glucose (5 mM, 10 mM, and 20 mM) alone for 48 h served as control. Significant level (p < 0.05) of reduced cell viability was observed in 12.5-µM butein-treated cells when compared to its corresponding control (glucose alone treated cells). Cell viability is further decreased (p < 0.001) while increasing the concentration of butein. Figure 3(b) shows phase-contrast microscopic picture of low (5 mM) and high (20 mM) concentration of glucose-treated cells. As seen in Figure 3(b), butein (25 µM)-treated cells show membrane blabbing and cell shrinkage. Butein alone (12.5 µM and 25 µM) treatment for 24 h did not show any statistical significance (data not shown) when compared with control cells.
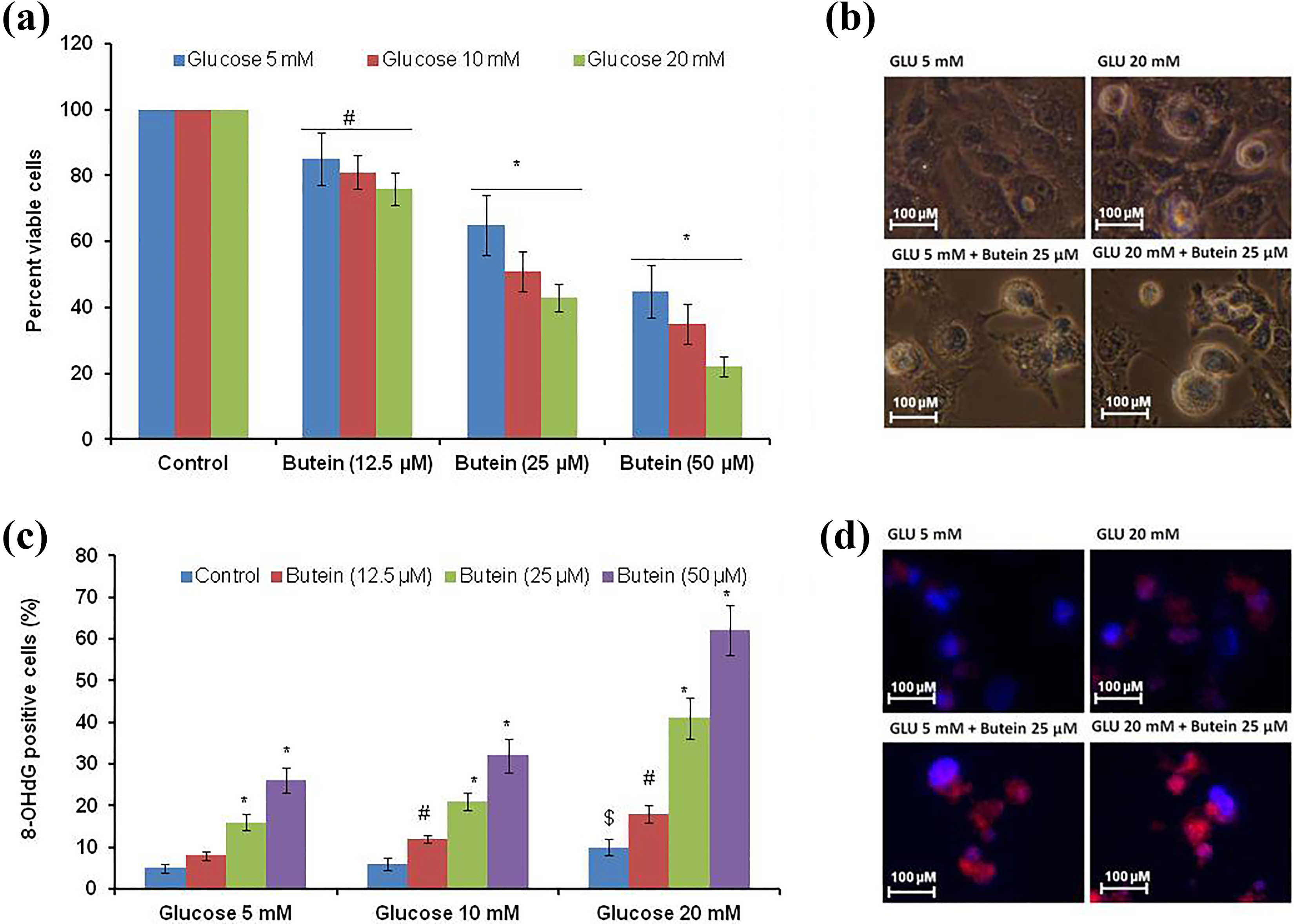
Cell viability (a and b) and oxidative DNA (c and d) damage in butein-treated cells. Cells were exposed to low (5 mM), medium (10 mM), and high (20 mM) concentrations of glucose for 48 h. Glucose-exposed cells (for 48 h) were incubated with butein (for 24 h). After the experimental period (72 h), cell viability (a) was analyzed. Experiments were repeated four times with triplicate in each time (n = 4). (b) The microscopic image (×40) of control and treated cells. 8-OHG levels were shown in (c). (d) Fluorescence image (×40) of 8-OHG. Values are expressed as mean ± SD. Statistical significance was calculated by one-way ANOVA with the Bonferroni multiple comparison posttest. #p < 0.05; *p < 0.001 versus respective control (different concentrations of glucose alone treated cells); $p < 0.05 versus 5-mM glucose alone treated cells. OHG: 8-hydroxy-2′-deoxyguanosine; SD: standard deviation; ANOVA: analysis of variance.
Figure 3(c) shows oxidative DNA damage in terms of 8-OHG in different experimental cells. We have noticed that high concentration of glucose (20 mM) induces significant level of 8-OHG (p < 0.05) in comparison to low (5 mM) glucose-treated cells. Exposure of cells with different concentrations of butein (12.5 µM, 25 µM, and 50 µM) was found to be raising the 8-OHG levels (p < 0.05; p < 0.001) when compared with the respective control cells. Figure 3(d) shows fluorescence microscope image of glucose (low and high) and butein (medium concentration) treated cells. Butein alone (12.5 and 25 µM) treatment for 24 h did not show any statistical significance (data not shown) when compared with control cells.
Next, we have examined the DNA strand breaks using comet analysis (Figure 4(a) to (c)). As seen in Figure 4(a), statistically significant (p < 0.05) tail length was observed in high level of glucose (20 mM) treated cells when compared with low level of glucose (5 mM)-treated cells. Butein (12.5 µM, 25 µM, and 50 µM) exposure was found to be inducing DNA strand breaks in low, medium, and high level of glucose-treated cells. We then analyzed the percent DNA present in tail (Figure 4(b)), which is consistent with DNA strand breaks. Figure 4(c) shows the fluorescence image of different experimental cells. No statistical significance was observed when butein alone (12.5 µM and 25 µM; data not shown)-treated cells compared with control cells.

Comet analysis. After the experimental period, comet analysis was performed. Experiments were repeated four times with triplicate in each time (n = 4). DNA strand breaks were measured in terms of tail length (a) and percent DNA in tail (b).(c) The respective fluorescence image (×60): “H” and “T”. One hundred cells from randomly selected 10 different fields were used for each slide. Values are expressed as mean ± SD. Statistical significance was calculated by one-way ANOVA with the Bonferroni multiple comparison posttest. *p < 0.001 versus respective control (different concentrations of glucose alone treated cells); $p < 0.05 versus 5-mM glucose alone treated cells. H: head region; T: tail; SD: standard deviation; ANOVA: analysis of variance.
We have observed a significant rise in 8-OHG and DNA damage in different experimental cells. We analyzed intracellular ROS (Figure 5(a) and (b)) and mitochondrial membrane potential (Figure 5(c) and (d)). Consistent with 8-OHG and comet analysis, high concentration of glucose (20 mM)-treated cells show significant increase (p < 0.05) in intracellular ROS and mitochondrial membrane potential in comparison to cells treated with low glucose (5 mM). Further increase (p < 0.05; p < 0.001) in intracellular ROS and mitochondrial membrane potential were observed in different concentrations of butein (25 µM and 50 µM)-treated cells when compared with respective control cells.

Intracellular ROS and mitochondrial membrane potential. (a) and (b) The intracellular ROS by DCF fluorescence dye (% control). (c) and (d) The mitochondrial membrane potential (fold change). Assays was performed after 72 h (glucose 48 h + butein 24 h = 72 h) of experimental period. Experiments were repeated four times with triplicate in each time (n = 4). Representative image of different experimental groups; green color indicates level of ROS (b), red color indicates intact mitochondria (d), decrease in red and increase in green indicates mitochondrial membrane damage (d), original magnification: 400×, Hoechst counter stain (blue) (b and d). Values are expressed as mean ± SD. Statistical significance was calculated by one-way ANOVA with the Bonferroni multiple comparison posttest. #p < 0.05; *p < 0.001 versus respective control (different concentrations of glucose alone treated cells); $p < 0.05 versus 5-mM glucose alone treated cells. ROS: reactive oxygen species; DCF: 2′,7′-dichlorofluorescein; SD: standard deviation; ANOVA: analysis of variance.
Changes of major antioxidant enzyme activities in glucose- and butein-treated cells
Table 1 presents the antioxidant enzyme activities such as CAT, SOD, GPx, and GR in different experimental cells. Glucose (20 mM; high concentration) decreases the activities (p < 0.05) of CAT, SOD, GPx, and GR in comparison to the 5-mM glucose-treated cells (low glucose). Low glucose-treated cells were exposed to 25-µM butein; significant decreased activities (p < 0.05) were observed in the abovementioned enzymes. However, when high glucose (20 mM)-treated cells were exposed to 25-µM butein, the activities of CAT, SOD, GPx, and GR were decreased at p < 0.001 level (when compared with low glucose) and p < 0.05 level (when compared with 20 mM glucose). These results indicate that butein decreased antioxidant enzyme activities irrespective of glucose concentration.
Activities of different antioxidant enzymes.a

SOD: superoxide dismutase; CAT: catalase; H2O2: hydrogen peroxide; GPx: glutathione peroxidase; GR: glutathione reductase; SD: standard deviation; ANOVA: analysis of variance.
aCells were treated as explained in materials and methods section. SOD-units/mg protein, CAT-pM of H2O2 utilized/mg protein, GPx-pM of Reduced glutathione (GSH) utilized/mg protein, and GR-pM of Nicotinamide adenine dinucleotide phosphate oxidase (NADPH) oxidized/min/mg protein. Values are mentioned as the mean ± SD (n = 4). Statistical significance were analyzed by one-way ANOVA with the Bonferroni multiple comparison posttest. “a” versus 5-mM glucose and “b” versus 20-mM glucose.
bp < 0.01.
cp < 0.05.
dp < 0.001.
Glucose on mROS
mROS levels were assayed in low glucose (5 mM)- and high glucose (20 mM)-treated cells (Figure 6). For this experiment, we have exposed the cells to low (5 mM) and high (20 mM) concentrations of glucose. It is observed that mROS is found to be increasing (p < 0.01) in high concentration of glucose-treated cells in comparison to low concentration of glucose-treated cells. For low concentration of glucose-treated cells exposed to butein (25 µM), mROS was found to be increased (p < 0.01) when compared with 5-mM glucose alone-treated cells. On the other hand, the cells treated with high concentration of glucose (20 mM) + butein (25 µM), the mROS levels were found to be further increasing (p < 0.01) in comparison to the cells that were treated with 20-mM glucose alone. This result indicates that butein acts as an anticancer agent through elevated mROS in glucose deprived as well as glucose well-fed cells.

Mitochondrial ROS levels in low and high glucose-exposed cells treated with butein for 24 h. Experiments were repeated four times with triplicate in each time (n = 4). Values are expressed as mean ± SD. Statistical significance was calculated by one-way ANOVA with the Bonferroni multiple comparison posttest. “a” versus low glucose (5 mM) and “b” versus high glucose (20 mM). #p < 0.05 and *p < 0.001. ROS: reactive oxygen species; SD: standard deviation; ANOVA: analysis of variance.
Effect of 2-DG on glucose-induced proliferation of NSCLCCs with or without butein
Figure 7(a) shows the effect of 2-DG on low, medium, and high glucose-treated cells. A significant level (p < 0.05) in inhibition of cell proliferation was observed in 2-DG (5 mM)-treated cells when compared to respective control cells. This inhibition further increased (p < 0.001) when 2-DG concentration was raised to 10 mM. We did not observe statistically significant level of inhibition when cells were treated with 2.5 mM of 2-DG. Pharmacological glycolysis inhibitor (2-DG) showed significant inhibition at 5-mM glucose-treated cells. So, we intended to test the effect of nontoxic concentration of 2-DG (2.5 mM) with the lowest concentration of butein used in this study (12.5 µM). Combination treatment of 2-DG (2.5 mM) + butein (12.5 µM) showed better cell growth inhibition than that of butein alone treatment (Figure 7(b)). This result indicates that 2-DG + butein have synergistic effect on glucose-induced proliferation of NSCLCCs.

Cell viability. (a) Cells were treated with either low or high glucose concentration with or without 2-DG for 24 h. (b) Synergistic effect of 2-DG and butein. Experiments were repeated four times with triplicate in each time (n = 4). Statistical significance was calculated by one-way ANOVA with the Bonferroni multiple comparison posttest. “a” versus low glucose (5 mM) and “b” versus high glucose (20 mM). #p < 0.05; @p < 0.01, and *p < 0.001. 2-DG: 2-deoxy glucose; ANOVA: analysis of variance.
Levels of p38 and ERK on glucose-induced proliferation
We have analyzed important signaling molecules involved in butein-induced cell death. As shown in Figure 8(a) and (b), butein increased p38 and ERK phosphorylation. We did not observe any change in c-Jun N-terminal kinase (JNK). Further to understand the mechanism of butein-induced cell death, we have treated the cells with p38 phosphorylation inhibitor SB 203580 (10 µmol/l) and with a well-known antioxidant such as NAC (5 mM). We have used these compounds individually as well as in combination. As seen in Figure 8(c), p38 phosphorylation inhibitor and NAC could completely inhibit the anticancer effect of butein, indicating the involvement of both these pathways.
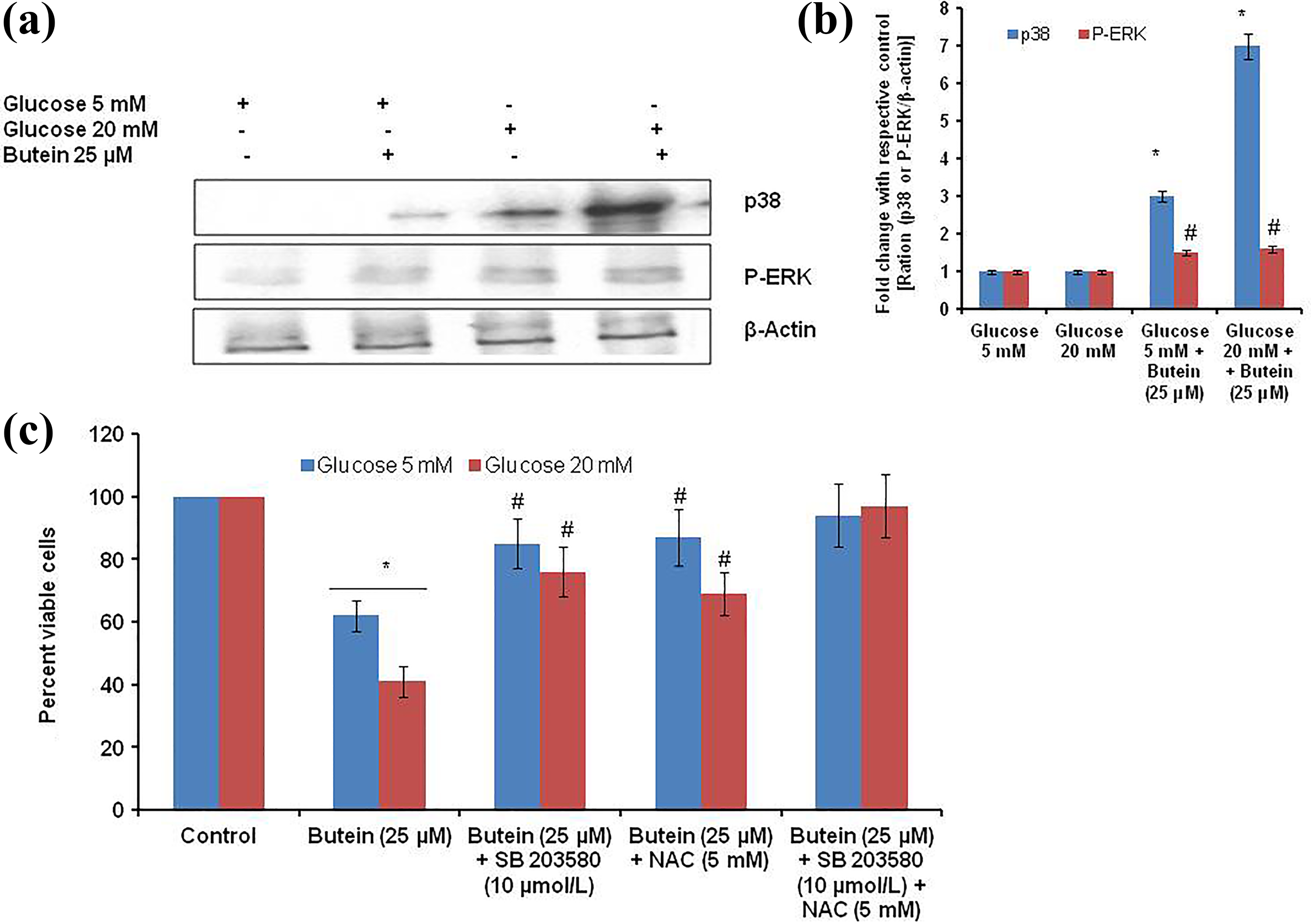
Levels of P-p38 & P-ERK, effect of NAC & p38 kinase inhibition in control, and experimental cells. (a) and (b) Level of protein expression by means of western blotting assay and densitometry ratio (respective protein/b-actin). Cell viability in glucose + butein-treated cells. Cells were pretreated with NAC (5 mM) and p38 kinase (SB203580; 10 µmol/l) for 30 min. Glucose + butein was then added and the incubation continued for 24 h. Cell viability (c) (MTT assay) was then analyzed. Statistical significance was calculated by one-way ANOVA with the Bonferroni multiple comparison posttest. “a” versus low glucose (5 mM) and “b” versus high glucose (20 mM). #p < 0.05 and *p < 0.001. ERK: extracellular signal-regulated kinases; NAC: N-acetylcysteine; ANOVA: analysis of variance; MTT: 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide.
Discussion
Canonical metabolic pathways are altered in pathological conditions, 34 which are most common in cancer cells. Growing cancer cells needs more energy, to meet their fast-growing requirements. 35 However, tumor niche does not have favorable environment for complete oxidative phosphorylation, meaning TCA cycle is reduced. On the other hand, glycolysis is increased, resulting in overgeneration of lactic acid levels in cancer cells. 36,37 In the above process, solid tumor tissue exposed gradient of glucose concentration, similar to that of oxygen levels. This tumor niche is responsible for various secondary complications of tumor, including drug resistance. 38 NSCLCCs show drug resistance against almost all the currently used chemotherapeutic drugs. 38 Mortality due to NSCLCCs is increasing day by day. Identifying new drug candidates to this cancer is of paramount importance of the hour. Secondary metabolites from plant sources show a remarkable efficacy against wide range of cancers. One such compound is butein. 16 –20 In the present study, we have tested the effect of butein against glucose deprived and glucose affluent conditions.
We found that NSCLCCs could not proliferate in completely glucose-deprived condition (0 mM) and also very high glucose condition (40 mM). So, we used three different concentrations of glucose such as low (5 mM), medium (10 mM), and high (20 mM). Low glucose (5 mM) used in this study is equal to that of 90 mg/dl, which falls in the normal blood glucose level. The high glucose (20 mM) used in this study is equal to that of 360 mg/dl, which comes under the glucose concentration in pathological conditions such as diabetes. Butein could very well be used as an anticancer agent in normal as well as diabetic subjects carrying cancer. NSCLCCs behave almost similar to that of thyroid cancer with respect to glucose concentration. 39 These three concentration-exposed cells were tested against three different concentrations of butein, that is, 12.5 µM, 25 µM, and 50 µM. The half maximal inhibitory concentration of butein in NSCLCCs was found to be 61.8 µM at 24 h. We have chosen the concentration butein based on the present and previously reported studies. 16,40 To our surprise, we observed that, irrespective of different concentrations of glucose, butein showed similar anticancer activity. In liver cancer cells, butein has shown its anticancer activity through the inhibition of HK II activity, 40 which is overexpressed in different solid tumors as well. Further, this enzyme is also overexpressed in anaerobic glycolysis. The observed anticancer effect could be due to inhibition of HK II inhibition in glucose-exposed cells.
Butein is known to have free radical quenching and antioxidant property. 21,41 To understand the anticancer effect of butein and its anticancer mechanism of action, we have analyzed oxidative stress by analyzing the oxidative DNA damage (strand breaks by comet analysis), 8-OHG, intracellular ROS, and mitochondrial membrane potential difference. All these results uniformly show that there was an increase in oxidative stress in butein-treated cells. As far as cancer cells are concerned, butein induces apoptosis-mediated cell death through the elevation of intracellular ROS. 42 Our present observation is in line with other investigators.
Primary antioxidant enzymes such as CAT, SOD, GPx, and GR are responsible for quenching increased intracellular ROS. These enzyme activities are decreased in high glucose (20 mM) alone-treated cells in comparison to that of low glucose alone-treated cells. These results indicate that glucose at higher (20 mM) concentration induces oxidative stress. These antioxidant enzymes were reduced in glucose-exposed cells (either low or high) that were incubated with butein. Increase in intracellular ROS and decreased activities of primary antioxidants enzymes correlates directly in butein-treated cells. Butein exert its anticancer activity through different mechanisms, which include induction of intracellular ROS. 16,21 We conclude that in NSCLCCs, butein inhibits cell proliferation through increased intracellular ROS.
Since butein has shown its anticancer effect in glucose-deprived and well-fed conditions, we then have analyzed the mROS in low glucose (5 mM)- and high glucose (20 mM)-treated cells. Twofold increase in mROS was observed in high glucose (20 mM) treated cells than that of low glucose-treated cells. It is known that hypoxia condition generates less mROS, whereas leak in electron transport chain complex I and complex II generate increased superoxide and hydrogen peroxide. An elevated mROS directs the cells for autophagy and apoptosis. 43 Even though we have observed 1.5-fold higher mROS in high amount of glucose-treated cells, we observe no cell death. It is known that cancer cells have resistant to apoptosis or autophagy-mediated cell death. 44 However, butein treatment further increases the mROS and causes cell proliferation inhibition in NSCLCCs. This is the first study to report that well-fed glucose (20 mM) could increase mROS in NSCLCCs than that of glucose-deprived cells (5 mM). Our studies have shown that an increase in intracellular ROS through butein caused DNA damage and G2/M arrest by activation of ataxia telangiectasia mutant pathway. 16,45
Pharmacological glycolysis inhibitor, 2-DG, dose dependently prevented glucose-mediated NSCLCCs proliferation. 46 This result gives way to target the glucose utilization in cancer cells. Next, we employed cancer cell sensitization through 2-DG, a glycolysis inhibitor. We have sensitized NSCLCCs with 2-DG (2.5 mM) and exposed them with minimal concentration of butein (12.5 µM). Glycolysis inhibition and plant-derived anticancer compound showed better effect in preventing NSCLCCs proliferation. One of the signaling molecules activated in butein-exposed cells is phosphorylate p38. 47 Our present finding correlates with the existing literature. Further, we and other investigators have also observed increased intracellular ROS and mROS. We have adopted SB 203580 to inhibit p38 phosphorylation and NAC to quench ROS. These compounds alone could not revert the butein-mediated inhibitory property of NSCLCCs. However, combination of these molecules shows a better effect in preventing butein-induced anticancer effect. These results indicate that both the mechanisms are involved in the anticancer effect of butein. Our findings directly correlate with findings of different investigators in different cancer cells.
In summary, this study throws the evidence that butein could be a promising anticancer agent irrespective of the cancer cell niche. Combination of 2-DG, a glycolysis inhibitor, and butein shows a synergistic anticancer effect against NSCLCCs.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.