
Abstract
High glucose (HG) induces vascular injury in diabetes. Hydroxysafflor yellow A (HSYA) has been used to ameliorate ischemic cardiovascular diseases in China for many years. In the present study, we assessed whether HSYA has a potential protective role in HG-induced human umbilical vein endothelial cell (HUVEC) injury. Cell viability was determined with an 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. Cell apoptosis was detected by fluorescein isothiocyanate/propidium iodide staining assay. The endothelial cell permeability was measured with a permeability assay. Cell adhesion molecule (CAM) expression, vascular endothelial growth factor, and basic fibroblast growth factor levels were detected with an enzyme-linked immunosorbent assay. Reactive oxygen species (ROS) formation was measured with a DCF-DA assay. Protein expression of NADPH oxidase 4 (NOX4) was measured by Western blotting. Our data indicated that HG increases HUVEC apoptosis, vascular permeability, monocyte adhesion, the level of CAMs, the formation of ROS, and NOX4 expression. Our data revealed that HG increases vascular injury, which is attenuated by HSYA. Because vascular inflammation has a key role in the development of diabetes mellitus, our results implied that HSYA is considered as a potential agent for diabetic vascular injury treatment.
Keywords
Introduction
Both obesity and physically inactive lifestyles are related to increases in the incidence of diabetes mellitus (DM), which is a major global health burden. 1,2 Diabetic vascular disease is one of the leading causes of death worldwide. 3,4 Extensive investigation has reported that DM impairs endothelial function via reducing vasodilatation and inhibiting endothelial cell (EC) repairs. 5,6 Previous studies have indicated that high glucose (HG) stimulates excessive reactive oxygen species (ROS) generation in many kinds of cells. 7 –9 Then, overexpression of ROS enhances cell dysfunction and apoptosis. 10,11 Thus, suppression of oxidative stress is considered as a potential strategy for DM. 12 It is important to find new therapy for treating HG-induced endothelial cell (EC) injury.
There are many studies that have demonstrated that hydroxysafflor yellow A (HSYA) has anti-inflammatory, 13 antioxidative, 14 and antiangiogenic functions both in vitro and in vivo. 15 HSYA is a key water-soluble compound of safflor yellow with a chalcone glycoside structure. HSYA has been isolated from Carthamus tinctorius 16 and is known as one of the standard elements for Carthami Flos quality control following the Chinese Pharmacopoeia, due to its high activity. HSYA has been applied widely to treat several diseases, including cardiovascular 17,18 and cerebrovascular diseases. 19 HSYA prevents oxidative stress generation, the inflammatory response, and cell apoptosis in spinal cord injury (SCI) rats, suggesting that HSYA may be a neuroprotective agent for SCI. 20 HSYA also suppresses idiopathic pulmonary fibrosis development via delaying lung fibroblast proliferation, migration, and differentiation. 21 In addition, HSYA reduces hepatocellular carcinoma (HCC) proliferation and metastasis via inhibiting angiogenesis. 22 However, the molecular mechanisms underlying HSYA protecting ECs remain largely unknown.
In this study, we aimed to investigate the potential effects of HSYA on HG-induced human umbilical vein endothelial cell (HUVEC) injury as well as the underlying mechanisms.
Materials and methods
Chemicals and reagents
Glucose was purchased from Sigma-Aldrich (St Louis, Missouri, USA). HSYA standards were obtained from China Pharmaceutical and Biological Products (Beijing, China). All antibodies, including primary and horseradish peroxidase-conjugated secondary antibodies, were purchased from Sigma-Aldrich.
Cell culture and treatment
HUVECs were purchased from American Type Culture Collection (ATCC), and cells were seeded in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, Massachusetts, USA) supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere of 5% carbon dioxide at 37°C.
The human monocytic cell lines THP-1 were purchased from ATCC and cultured in RPMI 1640 medium (Thermo Fisher Scientific) with 10% FBS, penicillin (100 units/ml), streptomycin (100 ng/ml), and 50 µM 2-ME (Fisher Scientific, Pittsburgh, Pennsylvania, USA) at 37°C in air atmosphere.
Cell viability
Cell viability was determined with a Cell Titer 96TM AQueous cell viability assay kit (MTS, Promega, Promega Corporation, Madison, WI). HUVECs (5 × 103 cells/well) were seeded in 96-well culture plates overnight. After the cells were treated with HSYA (0–50 μM) and glucose (50 μM) for 24 h, 20 μL CellTiter 96 Aqueous One Solution Reagent was added for 1 h at 37°C. The optical density (OD) was measured at 490 nm by a microplate reader (BioTek, Winooski, Vermont, USA).
Annexin-V/FITC assay
The apoptosis of HUVECs was quantified by an Annexin-V/fluorescein isothiocyanate (FITC) apoptosis assay kit (BD Biosciences, San Jose, California, USA) following the manufacturer’s instructions. The percentage of apoptotic cells was measured with a flow cytometer (BD, Minneapolis, MN, USA).
Permeability assay
The EC permeability was quantified by the flux of Evans blue-bound albumin across functional cell monolayers as previously described. HUVECs (5 × 104 cells/well) were seeded in a transwell for 24 h. Then, 0.5 mL of Evans blue solution was added to the upper chamber. The RPMI 1640 medium with 10% FBS was added to the lower chamber for 30 min. The OD of the lower chamber was evaluated at 650 nm with a microplate reader (BioTek).
Levels of CAMs with an ELISA
The levels of vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E-selectin were detected with an enzyme-linked immunosorbent assay (ELISA) (Sigma-Aldrich). Briefly, cells were treated with HSYA (0–50 μM) and glucose (50 μM) for 24 h and fixed in 1% paraformaldehyde. The mouse antihuman monoclonal antibodies were incubated for 1 h. Then, cells were washed with phosphate-buffered saline (PBS), treated with peroxidase-conjugated anti-mouse immunoglobulin G antibodies for 1 h, and the o-phenylenediamine substrate was added. All measurements were performed in triplicate.
HUVEC adhesion assay
The adherence of monocytes to ECs was detected by fluorescent labeling of monocytes, as previously described. 23 Monocytes were labeled with fluorescence-labeled using Calcein-AM (100 μM, R&D Systems). The monocytes and HUVECs were coincubated for 30 min at 37°C in air atmosphere. Nonadherent cells were washed with PBS. The OD was assessed at 492 nm (excitation) and 535 nm (emission) with a microplate reader (BioTek).
H2O2 release assay
The extracellular generation of hydrogen peroxide (H2O2) was tested with an Amplex Red Hydrogen Peroxide Assay Kit (Molecular Probes, Eugene, Oregon, USA) following the manufacturer’s protocol. Cells were resuspended in a Krebs–Ringer phosphate–glucose (KRPG) solution. The reaction mixture (100 μL, 50 μM Amplex Red reagent with 0.1 U/mL HRP in KRPG) was incubated at 37°C for 10 min. Then, KRPG solution (20 μL) was added to the cells for 30 min. The OD was assessed at 560 nm with a microplate reader (BioTek).
Intracellular ROS assay
The DCF-DA assay was used to measure the level of ROS. HUVECs were incubated with 2′,7′-dichloro dihydroflourescein diacetate (DCFH-DA) (10 μM) for 30 min at 37°C. The OD was evaluated at 485 nm (excitation) and 535 nm (emission) with a microplate reader (BioTek).
Western blotting
The total proteins were lysed using radioimmunoprecipitation assay (RIPA) buffer mixed with a protease inhibitor cocktail (Thermo Scientific, Rockford, Illinois, USA). The protein concentration was measured with a bicinchoninic acid assay (BCA) protein assay kit. Then, 30 µg of protein was separated with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (10%) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, Massachusetts, USA). The PVDF membranes were blocked with 5% nonfat milk for 1 h at room temperature. Then, the primary antibodies (anti-NOX4) were added to the membranes overnight at 4°C. The membranes were incubated with the secondary antibodies for 1 h at 37°C. Finally, the membranes were detected with an ECL reagent system (Thermo Scientific). The bands were analyzed with ImageJ Software (NIH, Bethesda, Maryland, USA).
VEGF and bFGF content in the cell culture supernatant by ELISA
The VEGF and bFGF levels were measured using a human VEGF ELISA kit (Thermo Fisher Scientific) and human bFGF ELISA kit (Thermo Fisher Scientific) following the manufacturer’s instructions. The OD was evaluated at 450 nm and 492 nm with a microplate reader (BioTek).
Statistical analysis
All measurements were performed in triplicate. All data were analyzed with SPSS 21.0 statistical software (SPSS, Inc., Chicago, Illinois, USA). The results are given as the mean ±standard deviation. Statistical significance was determined using a one-way analysis of variance. A p-value ≤0.05 was considered statistically significant.
Results
The effects of HSYA on HUVEC viability
To avoid possible effects of HSYA on HUVEC viability, cell viability was assessed with an MTS assay. The data indicated that HSYA had no effect on cell viability at a concentration of 50 µM (Figure 1(a)) for 72 h (Figure 1(b)). Therefore, a 50 µM concentration was used in all subsequent experiments.

Effect of HSYA and HG on the cell viability of HUVECs. (a) HUVECs were treated with different concentrations of HSYA (0–50 µM) for 24 h. Cell viability (OD490) was assessed with a MTS assay. (b) HUVECs were treated with different concentrations of HSYA (50 µM) for 0–72 h, and cell viability (OD490) was assessed with an MTS assay. (c) HUVECs were cultured with control or HG medium in the absence or presence of HSYA (50 µM), and cell viability was measured with an MTS assay. *p < 0.05 versus control group; #p < 0.05 versus HG group. HG: high glucose; HSYA: hydroxysafflor yellow A; HUVEC: HG-induced human umbilical vein endothelial cell; OD: optical density.
Compared with the control group, HUVEC viability in the HG group was decreased (p < 0.05, Figure 1(c)). HSYA-induced HUVEC viability was compared with the HG group for 24 h (p < 0.05, Figure 1(c)).
HSYA attenuates HG-induced HUVEC apoptosis
The percentages of apoptotic cells were quantified with an FITC/ propidium iodide (PI) and flow cytometer assay. After the cells were incubated with HG for 24 h, the rates of apoptotic cells were increased. This incremental apoptosis was reduced when the cells were treated with HSYA. Therefore, HSYA-reduced HUVEC apoptosis was compared with the HG group (Figure 2).

(a) HSYA reverses HG-induced HUVEC apoptosis. HUVECs were treated with HSYA (50 µM) and HG for 24 h, and (b) the rate of apoptosis in HUVECs was detected using an FITC/PI assay. The experiment was repeated at least three times. Data are shown as the mean ± SD of three experiments. *p < 0.05 versus control group. #p < 0.05 versus HG group. HG: high glucose; HSYA: hydroxysafflor yellow A; HUVEC: HG-induced human umbilical vein endothelial cell.
Effects of HSYA on HG-induced disruption of the endothelial barrier function in HUVECs
To investigate the effects of HG on EC permeability, a permeability assay was used. HG-induced EC permeability was monitored for 24 h (Figure 3(a)).

Effects of HSYA on HG-induced HUVEC permeability changes. (a) HUVECs were treated with HG for 24 h, and permeability was monitored. (b) HUVECs were treated with HSYA for 24 h, and permeability was monitored. (c) HUVECs were treated with HSYA and HG for 24 h, and permeability was monitored. The experiment was repeated at least three times. Data are shown as the mean ± SD of three experiments. *p < 0.05 versus control group. #p < 0.05 versus HG group. HG: high glucose; HSYA: hydroxysafflor yellow A; HUVEC: HG-induced human umbilical vein endothelial cell.
Next, we examined whether HSYA can alter HG-induced membrane disruption. HSYA (50 μM) did not alter the barrier integrity (Figure 3(b)). As shown in Figure 3(c), treatment with HSYA led to a decrease in HG-enhanced hyperpermeability.
Effects of HSYA on HG-enhanced CAM expression and THP-1 adhesion
To determine the effects of HG on the expression of CAM, an ELISA was used. HG resulted in an increase in CAM expression, including VCAM-1, ICAM-1, and E-selectin (Figure 4(a)). Further, inhibition of CAM expression was exhibited when HUVECs were treated with HSYA (Figure 4(a)).

Effects of HSYA on HG-induced inflammatory responses. (a) HG-induced CAM expression in HUVECs was determined. VCAM-1, ICAM-1, and E-selectin were detected with an ELISA. (b) The amounts of adherent human neutrophils were observed with a cell–cell adhesion assay. The experiment was repeated at least three times. Data are shown as the mean ± SD of three experiments. *p < 0.05 versus control group. #p < 0.05 versus HG group. HG: high glucose; HSYA: hydroxysafflor yellow A; HUVEC: HG-induced human umbilical vein endothelial cell; VCAM-1: vascular cell adhesion molecule-1; ICAM-1: intercellular adhesion molecule-1; CAM: cell adhesion molecule; ELISA: enzyme-linked immunosorbent assay.
To assess the effect of HSYA on the adhesion of monocytes to HUVECs, we detected the adhesion of THP-1 cells to HUVECs. Compared with the control group, HG enhanced adhesion ability. Treatment with HSYA led to a decrease in the adhesion of THP-1 cells to HG-induced HUVECs (Figure 4(b)).
Effects of HSYA on the level of VEGF and bFGF in the HG-treated HUVECs
Compared with the control group, the levels of VEGF (Figure 5(a)) and bFGF (Figure 5(b)) were enhanced in the HG group. Compared with the HG group, the levels of VEGF (Figure 5(a)) and bFGF (Figure 5(b)) in the HG + HSYA group were reduced.
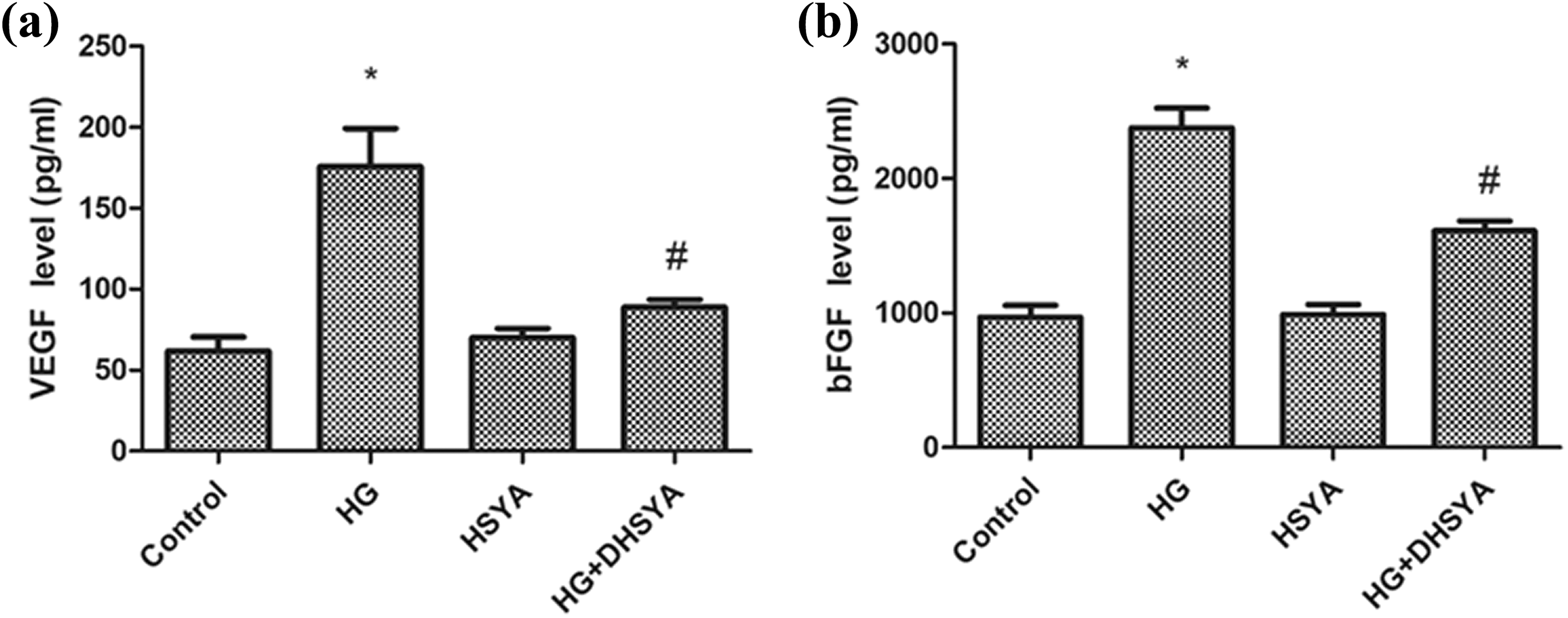
HSYA reverses the HG-increased levels of VEGF and bFGF in HUVECs. (a) VEGF levels in HUVECs were determined using an ELISA. (b) bFGF levels in HUVECs were determined with an ELISA. The experiment was repeated at least three times. Data are shown as the mean ± SD of three experiments. *p < 0.05 versus control group. #p < 0.05 versus HG group. HG: high glucose; HSYA: hydroxysafflor yellow A; HUVEC: HG-induced human umbilical vein endothelial cell; VCAM-1: vascular cell adhesion molecule-1; ICAM-1: intercellular adhesion molecule-1; CAM: cell adhesion molecule; ELISA: enzyme-linked immunosorbent assay.
Effect of HSYA on HG-induced oxidative stress
The H2O2 (Figure 6(a)) and ROS (Figure 6(b)) levels showed an increase after treatment with HG. However, HSYA had no effect on HUVEC oxidative stress, including H2O2 (Figure 6(a)) and ROS levels (Figure 6(b)). As shown in Figure 6, HSYA (50 μM) resulted in a suppression of HG-induced H2O2 and ROS levels.

Effects of HSYA on HG-induced H2O2 and ROS formation. Cells were treated with HSYA and HG for 24 h. (a) The production of H2O2 was detected with a H2O2 release assay. (b) The production of ROS was detected by DCFH-DA staining. The experiment was repeated at least three times. Data are shown as the mean ± SD of three experiments. *p < 0.05 versus control group. #p < 0.05 versus HG group. HG: high glucose; HSYA: hydroxysafflor yellow A; HUVEC: HG-induced human umbilical vein endothelial cell; ROS: reactive oxygen species; H2O2: hydrogen peroxide.
Effect of HG-induced ROS production by activating NOX4
Our data indicated that ROS levels were enhanced in HG-treated HUVECs. To determine the role of NOX on HG-induced oxidative stress, the NOX inhibitor DPI (2.5 μmol/L) was used. As shown in Figure 7(a), DPI decreased the level of ROS in response to HG. FITC/PI assays also showed that HUVEC apoptosis was reduced by DPI (Figure 7(b)).
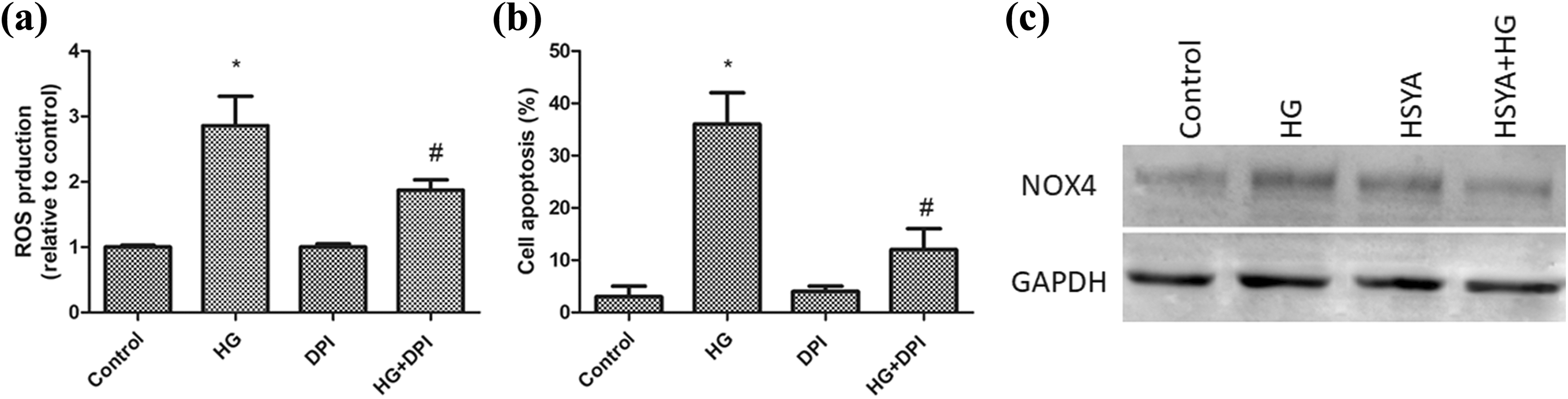
HSYA decreases HG-induced ROS generation and apoptosis in HUVECs through the NOX4 pathway. (a) DPI-decreased ROS production in HUVECs induced by HG. (b) DPI decreased the apoptosis rate in HG-treated HUVECs. (c) HSYA-decreased HG-induced NOX4 expression. Data are shown as the mean ± SD of three experiments. *p < 0.05 versus control group. #p < 0.05 versus HG group. HG: high glucose; HSYA: hydroxysafflor yellow A; HUVEC: HG-induced human umbilical vein endothelial cell; ROS: reactive oxygen species; NOX4: NADPH oxidase 4.
Effect of HSYA on HG-induced NOX4 activation
The NOX4 expression levels were increased in the HG-treated HUVECs (Figure 7(c)). In addition, HSYA inhibited HG-induced NOX4 expression (Figure 7(c)). These data suggested that HSYA inhibits HG-induced ROS levels in HUVECs via the NOX4 pathway.
Discussion
HG is a key factor responsible for ECECs injury, which subsequently leads to the development of diabetes. In this study, we showed that HSYA protects ECs against both oxidative stress and apoptosis. Furthermore, HG induces the expression of CAMs and promotes hyperpermeability and cell adhesion. HG enhances the levels of VEGF and bFGF in vitro. Interestingly, HSYA reverses all of these deteriorating effects of HG. Then, the potential mechanism for HSYA reducing HG-mediated EC injury was investigated. HSYA reduces the expression of NOX4, which is a key regulator of ROS generation in ECs. 24,25 Our data demonstrated that HSYA reduces HG-mediated ROS and apoptosis via the NOX4 pathway. These results indicated that HSYA might have a beneficial effect on diabetes treatment.
HUVECs are regarded as an in vitro model for the investigation of the role of ECs. 26 –28 Therefore, HUVECs were used to detect the effect of HSYA on ECs in this study. Increased monocyte-EC adhesion has been demonstrated in diabetes models both in vivo and in vitro. 29 It also has been reported that HG enhances leukocyte adhesion to ECs via upregulation of CAMs. In addition, CAMs are involved in diabetes-associated vascular complication development. 30 Our results confirmed that the expression of VCAM-1, ICAM-1, and E-selectin increases in HG-treated HUVECs. Our data are consistent with previous findings indicating that HG is a potent inductor of leukocyte adhesion to ECs via upregulation of E-selectin, ICAM-1, and VCAM-1. Additionally, the adhesion of monocytes to the endothelium is a key event in the vascular inflammation process. In this study, HSYA prevented the expression of CAMs and the adhesion of HUVECs. Based on the current data, HSYA inhibits HG-increased EC disruption. The past results showed that HSYA could inhibit methylglyoxal-induced injury in cultured human brain microvascular endothelial cells, which was associated with its antiglycation effect. 31 The in vitro data indicated that HSYA could increase both the angiogenesis and the keratinocyte migration. HSYA suppressed the nitric oxide production, demonstrating the anti-inflammatory role of HSYA. The in vivo data showed that HSYA improved the rate of wound closure. Furthermore, HSYA increased the collagen content, VEGF, and TGF-β1 expression. Therefore, HSYA should be considered a new therapeutic strategy in the treatment of diabetes. 32
VEGF is the most efficient cytokine for stimulating EC viability, and it has many biological effects including elevating vascular permeability and inflammatory responses. 33 bFGF is also related to inflammatory responses. 34 We found that HG reduces VEGF and bFGF, which is reversed by HSYA. These data suggested that HSYA decreases HG and triggers vascular permeability and inflammatory responses via reducing VEGF and bFGF levels. Therefore, HSYA may be used in vascular inflammatory disease treatment.
Previous studies have indicated that HG prompts oxidative stress and ROS generation. 35 We have detected the protective effect of HSYA on HG-induced cellular H2O2 concentrations and oxidative stress. HSYA mitigated HG-induced cellular H2O2 content and ROS in HUVECs. NOX4 is the key regulator in EC dysfunction. 36 We found that HG-induced ROS levels in HUVECs are reduced by DPI. Further, HG-induced HUVEC apoptosis is also reduced by DPI. The data suggested that NOX4 plays a key role in ROS generation and cell apoptosis induced by HG. In addition, HSYA inhibits HG-induced ROS levels in HUVECs via the NOX4 pathway.
In summary, our results demonstrated that HSYA has potent therapeutic benefits against HG-induced EC injury via attenuation of H2O2 and ROS generation, NOX4 activation, adhesion molecules and monocyte EC adhesion. Our findings indicated that HSYA may be considered as a potent agent for diabetic vascular inflammatory disease treatment. Although HYSA has various bioactivity effects, it is currently mainly used in cardiovascular and cerebrovascular disease treatment. More clinical and translational research studies are needed, especially for diabetic vascular inflammatory disease, as well as studies focusing on the molecular mechanisms of HYSA. Further research on the molecular mechanisms of HYSA is needed, and the number of clinical trials of HYSA needs to be increased.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Discipline Construction Project of Health and Family Planning Commission of Pudong New Area (nos. PWZxq2017-17, PWZxk2017-07, and PWZzb2017-22).