
Abstract
Carisoprodol is a widely prescribed muscle relaxant and is also a drug known to be a subject to abuse. Despite the fact that carisoprodol has been available for prescription since 1959, a number of gaps in our knowledge of the toxicokinetics of this common drug exist. For example, the volume of distribution (Vd) for carisoprodol in humans has not been reported. A two-compartment pharmacokinetic model describing carisoprodol metabolism and that of the primary metabolite, meprobamate, was developed to better understand the pharmacokinetics of this drug. The model accounts for first pass metabolism of carisoprodol and was able to replicate the data from several previously reported data sets. Based on an analysis of four different data sets, the Vd for carisoprodol ranged from 0.93 to 1.3 L/kg, while that for meprobamate ranged from 1.4 to 1.6 L/kg. The model was also used to estimate the probable dose of this drug in an individual where questions concerning the drug’s role in her death had been posed. The model may, therefore, have significant utility for estimating doses of carisoprodol in medicolegal cases.
Introduction
Carisoprodol (RS-2-{[(aminocarbonyl)oxy]methyl}-2-methylpentyl isopropyl carbamate, marketed as SOMA, Somadril, MioRelax) is a widely prescribed skeletal muscle relaxant, particularly used for managing lower back pain. 1,2 First introduced in 1959, it is indicated “as an adjunct to rest, physical therapy, and other measures for the relief of discomfort associated with acute, painful musculoskeletal conditions.” 3 In 2013, approximately 8.5 million “carisoprodol products” were dispensed in the United States. 4 The principal metabolite of carisoprodol via delakylation of the carbamate amine is another drug with similar action, meprobamate. 5 Both drugs exert depressant effects on the central nervous system.
Carisoprodol is a drug of abuse and is commonly noted to be a complicating factor in accidental deaths, in particular those involving motor vehicles. 1,6 –11 Data from the United States in 2013 indicate that approximately 3.7 million individuals reported using carisoprodol at least once for nonmedical use in their lifetime. 4 Bramness reported that carisoprodol ranks 17th among commonly encountered drugs in the US emergency room overdose cases. 12 Studies show that after carisoprodol was more tightly regulated in Norway in 2007, the detection of this drug in motor vehicle death cases declined. 13 As a result of its abuse potential, there have been numerous calls to ban or tighten regulation of this drug 14 and the drug was classified as a schedule IV drug (“low potential for abuse and low risk of dependence”) by the US government in 2012.
The published data on carisoprodol pharmacokinetics are incomplete as is often the case for drugs introduced many decades in the past. Once ingested, carisoprodol has a very rapid onset of action (generally within 30 min) with the muscle relaxing and analgesic effects lasting 4–6 h. At typical doses, serum concentrations reach a peak at about 1 h and then rapidly diminish as the drug is converted to meprobamate with a half-life estimated at approximately 100 min. 15 Meprobamate is metabolized much more slowly, with a half-life reported to range from 6 to 17 h. 5 The ratio of the two drugs in plasma therefore gives an indication of the dosing time frame (assuming that only carisoprodol is taken, not a combination of the two). The volume of distribution (Vd) for meprobamate is reportedly 0.7 L/kg, although the basis for this value is uncertain. 5 No Vd value for carisoprodol is reported. Analysis of an animal study conducted by the US National Toxicology program yields a Vd estimate for carisoprodol of 0.72 L/kg in rats and 0.65 L/kg in mice. 16 After death, carisoprodol is reported to undergo insignificant postmortem redistribution (PMR). 17
Metabolism of carisoprodol to meprobamate occurs primarily via dealkylation by CYP2C19 in the liver. 18 Meprobamate is subsequently hydrolyzed to hydroxymeprobamate prior to urinary excretion. Studies have indicated that CYP2C19 polymorphism contributes to variations in carisoprodol metabolism in humans. 1 The frequency of the poor metabolizer phenotype (−/−) varies according to ancestry, with an occurrence of approximately 4% in population groups of European and African ancestry, 1% in populations of Mexican ancestry, approximately 20% in populations of Asian or Southeast Asian ancestry, and up to 70% in Pacific Islanders. 19,20 The heterozygous phenotype (+/−) is more common but does not appear to markedly affect carisoprodol metabolism compared to the (+/+) efficient metabolizers. 1
Because there are gaps in knowledge concerning the pharmacokinetics of carisoprodol (in particular the Vd), a model was developed to predict the disposition of this drug and its metabolite in humans. The model was subsequently used to evaluate a medicolegal case in which the probable carisoprodol intake of the individual was a subject of interest.
Methods
To predict carisoprodol and meprobamate concentrations in response to a specific oral dosing pattern, a model was developed to describe the metabolic situation shown in Figure 1. After ingestion of an oral dose of carisoprodol, the drug is absorbed across the walls of the gastrointestinal tract and is transported to the liver prior to entering the general blood circulation. The model therefore accounts for first pass metabolism of carisoprodol, an important factor in this rapidly metabolized drug. In the liver, carisoprodol is metabolized to meprobamate. Meprobamate, in turn, is metabolized in the liver to hydroxymeprobamate, which is eliminated primarily via the urine. 21 The model therefore has two compartments, the liver and blood. Given the relatively simple kinetics of this system, use of a compartmental model appeared sufficient and a full physiologically based pharmacokinetic model was not considered necessary.
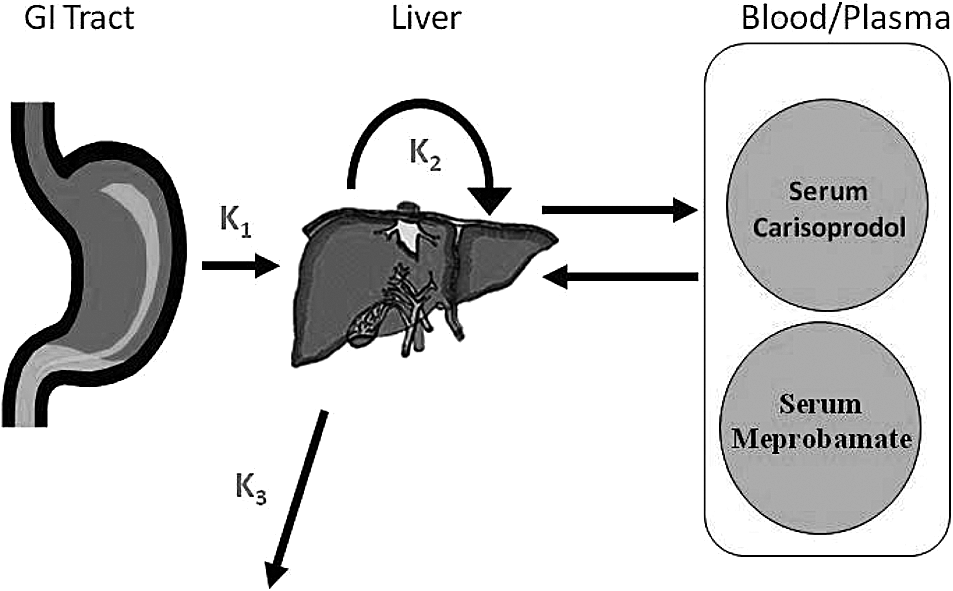
Schematic of PBPK model for carisoprodol metabolism indicating the three key compartments. PBPK: physiologically based pharmacokinetic.
The ordinary differentially equations used in the model were adapted from the model of Taft et al. which described the metabolism of the drug venlafaxine. 22 Each of the critical metabolic steps (i.e. absorption of carisoprodol, metabolism of carisoprodol to meprobamate, elimination of meprobamate) is described by rate constants (designated as k1, k2, and k3, respectively, as shown in Figure 1). Transport between compartments is limited by hepatic blood flow. The resulting model code was solved using Berkeley Madonna (Macey and Oster, Berkeley, California, USA). The initial model parameters were set based on the values obtained from the published literature or, where data were lacking, professional judgment. For the Vd of carisoprodol, the rat value obtained from the National Toxicology Program study was used as a starting point. Initial parameter values were then optimized using Berkeley Madonna’s curve fitting function using pharmacokinetic data reported in four different clinical studies. The curve fit function searches for input value combinations that give the lowest root mean square error between the observed literature data and the model predictions. All four studies (Olsen et al., Kucharczyk et al., Bramness et al., and Simon et al.) involved single doses of carisoprodol (ranging from 250 to 700 mg) administered to healthy adult volunteers with blood concentrations monitored for at least several hours. 1,15,23,24 The study by Kucharczyk et al. measured the plasma levels of carisoprodol for 8 h after a 350 mg oral dose and the studies by Olsen et al. and Bramness et al. measured the serum levels of both carisoprodol and meprobamate after a 700 mg oral carisoprodol dose (24 and 12 h, respectively), while the study by Simon et al. evaluated the kinetics of both chemicals (carisoprodol doses 250 or 350 mg) over 48 h. A limitation of the first three studies is a lack of data on body weight; for these studies population average values from literature sources were used. Model input values are given in Table 1 and the final model code is given in Appendix 1.
Input parameters and half-life predictions for the PBPK model.

PBPK: physiologically based pharmacokinetic; USEPA: United States Environmental Protection Agency.
aKucharczyk et al. only evaluated carisoprodol kinetics.
bBased on USEPA, 1997. 28 For the data of Bramness et al., the value was taken from Droyvold et al. 25
cFrom Taft et al. 22
dSubjects were fasted in the studies by Kucharczyk and Olsen et al., and not fasted in other studies.
eValues in parentheses are those reported by the study authors. In some cases, values are not reported (NA).
The final model was then used to predict concentrations of carisoprodol and meprobamate in a woman who died from a presumed multidrug overdose while under treatment for bone cancer. Levels of carisoprodol and meprobamate in the woman’s blood at autopsy were 22.8 and 30.8 mg/L, respectively. The goal was to determine whether these autopsy concentrations could be obtained if taking the drug at the prescribed level, or if not, whether they were consistent with a substantial overdose. The model inputs used to fit the data of Simon et al. (the most comprehensive data set with actual body weight information) were used as inputs for the model with the exception of the woman’s specific body weight. Because the woman was of Caucasian ethnicity, she was assumed to be an efficient metabolizer of carisoprodol.
Results
Fits of the model to empirical study data are shown in Figure 2(a) to (d). Although there were slight differences in the model input values required to fit the four different data sets, for most parameters, these differences were small and not unexpected, given intraindividual and inter-study variability. The estimated Vd of carisoprodol ranged from 0.93 to 1.26, while the estimates of the Vd of meprobamate ranged from 1.4 to 1.6, higher than the 0.7 L/kg value suggested by Robertson and Marinetti. 5 The rate constants for carisoprodol and meprobamate metabolism were generally similar among the four data sets. Predicted values of half-life (t 1/.2) for the two drugs were similar to those reported in the actual studies. In order to obtain optimal fit, the body weight used with the data set of Bramness et al. was adjusted from 70 to 75 kg, based on the authors’ statement that the subjects were young adults and taking into recent body weight data for this group in Norway, the country where the study was conducted. 25

Fit of PBPK model predictions to data of (a) Kucharczyk et al., (b) Olsen et al., (c) Simon et al., and (d) Bramness et al. Data for the poor metabolizer phenotype in Bramness et al. are shown for comparison but were not modeled. Error bars for Kucharczyk et al. are SEM and for Olsen et al. are 1 SE. Measurement error was not indicated by the other study authors. Data for the 250 mg dose in Simon et al. was multiplied by 1.4 to allow graphing with the 350 mg dose. PBPK: physiologically based pharmacokinetic; SEM: standard error of mean; SE: standard error.
For the medicolegal case, Figure 3 shows the predicted concentrations of carisoprodol and meprobamate in blood resulting from consumption of four daily carisoprodol doses consisting of either single 350 mg tablets (the typical dosing pattern) or five tablets per dose (1750 mg). The latter could be typical of a chronic abuse situation. Under the typical dosing situation, concentrations of carisoprodol never exceed 2.4 mg/L, almost 10 times less than the level found in the individual’s blood at autopsy. Thus, the individual could not have been following the recommended dosing schedule. Even a dose five times higher than the recommended level would not produce the concentrations of carisoprodol and meprobamate found at autopsy due to the relatively fast elimination of carisoprodol relative to meprobamate. As shown in Figure 3, while such a dose produces plausible concentrations of the parent, concentrations of the metabolite are far overpredicted. A more likely explanation for the concentrations observed was found to be a substantial overdose taken shortly before death (Figure 4). Only in this scenario do the individual’s predicted carisoprodol and meprobamate concentrations approximate the concentration and time relationship actually observed.
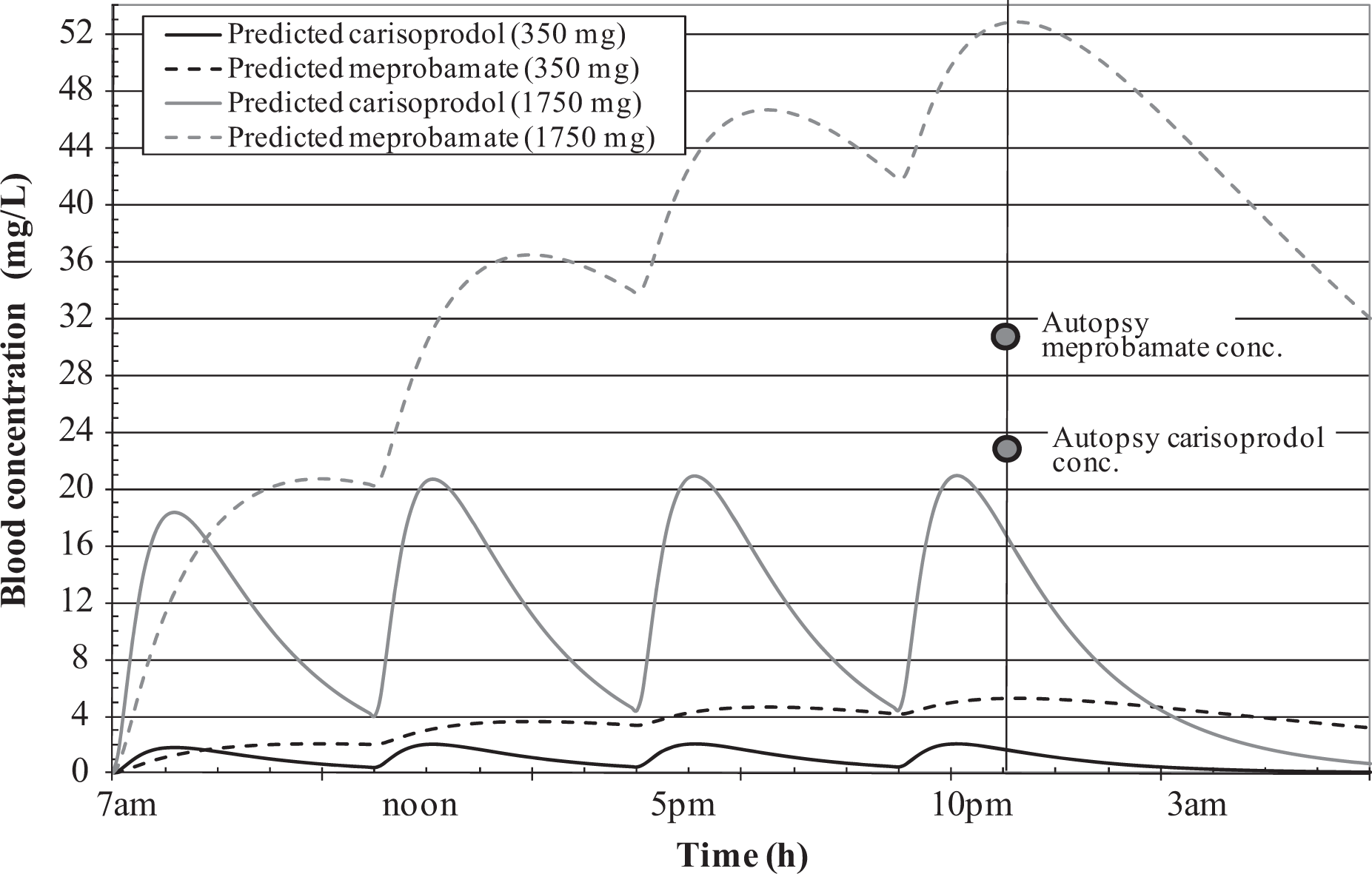
PBPK model predictions for case study assuming four regular doses of 350 or 1750 mg/day. Blood concentrations of carisoprodol and meprobamate measured at autopsy are indicated in the shaded circles. A regular dosing pattern does not result in concentrations that match those observed. PBPK: physiologically based pharmacokinetic.

PBPK model predictions for case study assuming three regular doses followed by five times a 250 mg dose at 10 p.m. Blood concentrations of carisoprodol and meprobamate measured at autopsy are indicated in the shaded circles. This scenario provides a better match to the observed concentrations than the scenarios described in Figure 3. PBPK: physiologically based pharmacokinetic.
Discussion
The work described above illustrates the utility of simple modeling in understanding particular aspects of drug disposition. By fitting a model to the results of several published data sets, values for the Vd of carisoprodol and meprobamate were established to address the lack of such values in the reported literature. The work also illustrates the value of modeling in exploring potential dosing scenarios in cases where dosing patterns are unknown but of interest from a medicolegal standpoint.
The values obtained for the Vd for carisoprodol are somewhat different from those that can be obtained from studies in rats and mice (0.72 and 0.65 L/kg, respectively), 16 although three of the clinical studies are in close agreement, with an average Vd value (i.e. 0.94 L/kg) that may not be inconsistent with the rodent values when interspecies variation and differences in methodology are considered (i.e. IV vs. oral dosing). The values obtained for the Vd of meprobamate (average 1.5 L/kg) are substantially above the 0.7 L/kg reported by Robertson and Marinetti. 5 However, the latter is given in a review article with no reference to primary data or the method by which it was determined. No set of input values were found which would allow the model to fit the three sets of clinical data using 0.7 L/kg as the Vd for meprobamate.
An attempt was also made to fit the poor metabolizer data of Bramness et al. (shown in Figure 2(d)), although this was not possible with the current model structure. Interestingly, to obtain a reasonable fit, a biphasic carisoprodol absorption curve was required, with a much lower rate of uptake in the middle portion of the curve. No good biological support for this apparent discontinuity in carisoprodol absorption into blood could be found and thus modeling it seemed questionable. Bramness et al. noted this phenomenon and stated it “may be an indication of a certain first pass metabolism of the drug.” 1 However, if it were simply a matter of reduced carisoprodol metabolic capacity in poor metabolizers across the entire dose range, the entire curve should be shifted upward and to the left without the notable change in slope near the top.
The four data sets examined considered doses that were in the normal therapeutic range. It is true that even among those with the efficient metabolizer phenotype, the metabolic capacity for carisoprodol may be substantially saturated at very large doses. For example, Goldberg showed that the half-life of carisoprodol in an individual consuming an overdose of twenty-seven 350 mg carisoprodol tablets (most of which may have been vomited) was on the order of 15 h versus the standard value of 100 min. 26 Clearly in such a case, the normal processes of drug metabolism are saturated and the metabolism shifts to a zero-order state. In such a situation, the reduced metabolism of carisoprodol to meprobamate would result in blood carisoprodol concentrations that are higher than blood meprobamate concentrations. 27 Because this inversion of the normal carisoprodol/meprobamate concentration ratio was not apparent in the medicolegal case described, it seems unlikely that saturation of metabolism played a significant role.
In any case involving the evaluation of autopsy drug measurement data, it is important to take into account the phenomenon of PMR. PMR involves the migration of chemicals partitioned into the tissues back into the blood as those tissue break down after death. PMR acts to inflate the concentrations of drugs observed in blood at autopsy relative to those that were present when the individual died. PMR is most significant for chemicals that are highly fat soluble and/or have extensive tissue distribution (i.e. high volumes of distribution). Unlike many drugs involved in fatalities (e.g. opioids), the Vd of carisoprodol and meprobamate is fairly low, and thus these compounds would not be expected to undergo significant PMR. 17
In summary, a two-compartment pharmacokinetic model describing carisoprodol metabolism and that of the primary metabolite, meprobamate, was developed to better understand the pharmacokinetics of this drug. Based on an analysis of the data, the volumes of distribution for carisoprodol and meprobamate were determined, which help to fill a data gap in the published literature. The model was also used successfully to estimate the probable dose of this drug in an individual where questions concerning the drug’s role in her death had been posed. The model may, therefore, have significant utility for estimating doses of carisoprodol in medicolegal cases.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The initial stages of this research were funded in connection with a legal case involving a wrongful death allegation associated with carisoprodol use.