
Abstract
Glyceryl trinitrate (GTN) has been used widely as a potent vasodilator to treat heart conditions, such as angina pectoris and chronic heart failure. This study aims to elucidate the effect of exogenous nitric oxide (NO) administration, using GTN, on carbon tetrachloride (CCl4)-induced oxidative stress and liver injury in rats. The results obtained demonstrated that NO generated by the administration of GTN affords protection against CCl4-induced oxidative stress and liver injury. Administration of CCl4 resulted in a significant (p < 0.001) increase in lipid peroxidation and tissue damage markers (aspartate and alanine transaminase and lactate dehydrogenase) release in serum. Parallel to these changes, CCl4 also caused downregulation of antioxidant enzymes including glutathione peroxidase (GPx), glutathione reductase (GR), and glutathione-S-transferase (GST), and several fold induction in γ-glutamyl transpeptidase (GGT) activity. Subsequent administration of GTN resulted in significant (p < 0.001) recovery of GSH-metabolizing enzymes in a dose-dependent manner. Further, administration of NO inhibitor, NG-nitro-
Introduction
One of the fundamental mechanisms of cell biology is defense against oxidative stress. Evidence indicate that reactive oxygen intermediates are pathogenic in several types of liver injury. 1 In most of the disease conditions and aging, the rate of generation of reactive oxygen species exceeds that of their removal, which leads to oxidative stress and depletion of endogenous antioxidants. Since, oxidative stress is an important contributor to the aging process and many diseases, the focus must be on reducing the oxidative stress in vivo. 2
Carbon tetrachloride (CCl4) is a well-known hepatotoxicant that causes oxidative stress-mediated liver injury and has similar mechanism in animals and humans. 1 CCl4 toxicity results from its bioactivation to the highly toxic reactive trichloromethyl peroxyl radical that subsequently attack the polyunsaturated fatty acids of membrane lipids to propagate a chain reaction resulting in peroxidative decomposition of cytoplasmic membrane lipids, 3 –6 leading to progression of liver damage with subsequent hepatocellular carcinoma. 7 –9 In our previous study, we have shown the role of plant extract as an antioxidant in attenuating CCl4-induced hepatotoxicity by inhibiting lipid peroxidation (LPO) and increasing antioxidant enzymes activity. 10
Earlier studies suggest that nitric oxide (NO) may act as an antioxidant and may interact with superoxide anion and other radicals to produce less toxic species. 11 –14 In contrast, other evidence suggests that NO may interact with reactive oxygen intermediates to form more toxic species. 15 Glyceryl trinitrate (GTN) functions as an endogenous physiological mediator by its bioconversion to vasoactive NO. 16 Recent studies have shown that GTN can improve the hepatocyte engraftment and correction of metabolic disease in knockout mice, and GTN therapy is postulated to be beneficial in hepatocyte transplantation for the treatment of patients with liver diseases. 17 Previous studies have shown that exogenous administration of NO donors decreases oxidant-mediated hepatotoxicity in hepatocyte tissue culture model, in vivo endotoxin-mediated hepatic injury, and inhibition of NO production potentiates cellular damage. 18,19 On one hand, NO may act as a scavenger for reactive oxygen species, 20 while reacting with carbon-centered alkoxyl and peroxyl radicals at a rapid rate to terminate LPO, on the other hand excess NO production can cause oxidative damage to membrane lipids, DNA, proteins, and lipoproteins. 21 –24 Controversial issue remains whether the augmented production of NO serves a protective or deleterious role in the liver. The aim of this study was to study the potential effect of exogenous NO donor and inhibitor on acute liver damage caused by CCl4 administration in rats.
Materials and methods
Chemicals
Glutathione reductase (GR), tris-hydrochloric acid (HCl), thiobarbituric acid, oxidized and reduced GSH, reduced form of nicotinamide adenine dinucleotide phosphate (NADPH), 2-mercaptoethanol, dithiothreitol, phenylmethylsulfonyl fluoride, pyridoxal-5-phosphate, glucose-6-phosphate, 1-chloro-2,4-dinitrobenzene (CDNB), 5,5′-dithio-bis-2-nitrobenzoic acid, NG-nitro-
Preparation of CCl4
The CCl4 solution was prepared by the method followed by Lindroos et al. 24
Animals and treatment
Wistar albino male rats (4–6 weeks old) weighing 125–150 g used in this study were housed in an air-conditioned room and had free access to pellet diet and water ad libitum. Approval was obtained from the ethics committee of the Hamdard University, New Delhi, India.
Experimental design
For studying the effect of GTN on CCl4-mediated induction of hepatic toxicity, 42 Wistar albino male rats weighing 100–150 g were taken and divided into seven groups of six rats per group. All the animals received intraperitoneal injections, and were killed 24 h after the last treatment. The dose selection for GTN, Group I received saline (0.85% sodium chloride (NaCl)). Group II received GTN (6 mg kg−1 body weight (b. wt)). Group III received Group IV received CCl4 (1 mL kg−1 b. wt in corn oil). Group V received CCl4 (1 mL kg−1 b. wt in corn oil) + GTN (3 mg kg−1 b. wt after 1 h of CCl4 administration). Group VI received CCl4 (1 mL kg−1 b. wt in corn oil) + GTN (6 mg kg−1 b. wt after 1 h of CCl4 administration). Group VII received CCl4 (1 mL kg−1 b. wt in corn oil) +
Just before the killing, blood samples from retro-orbital sinus of these animals were collected for the estimation of serum transaminases (alanine transaminase (ALT), aspartate aminotransferase (AST) lactate dehydrogenase (LDH), and γ-glutamyl transpeptidase (GGT). Livers were collected and processed for the estimation of antioxidants enzymes GR, glutathione S-transferase (GST), and glutathione peroxidase (GPx) as well as for histopathological studies.
Preparation of postmitochondrial supernatant and microsome
Livers were quickly removed, perfused immediately with ice-cold saline (0.85% w/v NaCl) and homogenized in chilled phosphate buffer (0.1 M, pH 7.4) that contained potassium chloride (KCl; 1.17% w/v) using a homogenizer. The homogenate was filtered through a muslin cloth and was centrifuged at 800 × g for 5 min at 4°C to separate the nuclear debris. The aliquot obtained was centrifuged at 1,05,000 × g for 20 min at 4°C to obtain postmitochondrial supernatant (PMS), which was used as a source of enzymes. A portion of the PMS was centrifuged in an ultracentrifuge, and the pellet was considered to be the microsomal fraction and was suspended in phosphate buffer (0.1 M, pH 7.4) containing KCl (1.17% w/v).
Lipid peroxidation
The assay for hepatic microsomal LPO was done following the method of Wright et al. 28 The reaction mixture, in a total volume of 1.0 mL, contained 0.58 mL phosphate buffer (0.1 M, pH 7.4), 0.2 mL microsome (10% w/v), 0.2 mL ascorbic acid (100 mM), and 0.02 mL ferric chloride (100 mM). The reaction mixture was incubated at 37°C in a shaking water bath for 1 h. The reaction was stopped by the addition of 1.0 mL trichloroacetic acid (TCA 10% w/v). Following addition of 1.0 mL thiobarbituric acid (0.67% w/v), all tubes were placed in a boiling water bath for a period of 20 min. At the end, the tubes were shifted to an ice bath and centrifuged at 2500 × g for 10 min. The amount of malondialdehyde (MDA) formed in each of the sample was assessed by measuring the optical density of the supernatant at 535 nm using a spectrophotometer against a reagent blank. The results were expressed as nanomoles of MDA formed per hour per gram tissue at 37°C using a molar extinction coefficient of 1.56 × 105 M−1 cm−1.
GST activity
GST activity was measured by the method of Habig et al. 29 The reaction mixture consisted of 1.425 mL phosphate buffer (0.1 M, pH 6.5), 0.2 mL reduced GSH (1.0 mM), 0.025 mL CDNB (1.0 mM), and 0.34 mL of PMS (10% w/v), with a total volume of 2.0 mL. The changes in absorbance were recorded at 340 nm and enzyme activity was calculated as nanomole CDNB conjugate formed per minute per milligram protein using a molar extinction coefficient of 9.63 × 103 M−1 cm−1.
GR activity
GR activity was assayed by the method of Carlberg and Mannervik. 30 The assay system consisted of 1.65 mL phosphate buffer (0.1 M, pH 7.6), 0.1 mL ethylene diamine-tetra acetic acid (EDTA; 0.5 mM), 0.05 mL oxidized GSH (1.0 mM), 0.1 mL NADPH (0.1 mM), and 0.1 mL of PMS (10% w/v) in a total volume of 2.0 mL. The enzyme activity was quantitated at 25°C by measuring the disappearance of NADPH at 340 nm and was calculated as nanomoles of NADPH oxidized per minute per milligram protein using a molar extinction coefficient of 6.22 × 103 M−1 cm−1.
GPx activity
GPx activity was measured according to the procedure described by Mohandas et al. 31 The reaction mixture consisted of 1.44 mL 0.05 M phosphate buffer, pH 7.0, 0.1 mL 1.0 mM EDTA, 0.10 mL 1.0 mM sodium azide, 0.05 mL 1 U mL−1 GSH reductase, 0.10 mL 1.0 mM GSH, 0.10 mL 2 mM NADPH, 0.01 mL 0.25 mM hydrogen peroxide (H2O2), and 0.10 mL 10% PMS in a total volume of 2.0 mL. Disappearance of NADPH at 340 nm was recorded at 25°C. Enzyme activity was calculated as nanomoles of NADPH oxidized per minute per milligram protein using a molar extinction coefficient of 6.22 × 103 M−1 cm−1.
GGT activity
The GGT activity was determined by the method of Orlowski and Meister 32 using γ-glutamyl-p-nitroanilide as a substrate. The reaction mixture was in a total volume of 1.0 mL and contained 0.2 mL homogenate (10%w/v), which was incubated with 0.8 mL of the substrate mixture (containing 4 mM γ-glutamyl-p-nitroanilide, 40 mM glycylglycine, and 11 mM magnesium chloride in 185 mM tris-HCl buffer, pH 8.25) at 37°C. Ten minutes after the initiation of the reaction, 1.0 mL of TCA (25% w/v) was added and mixed to terminate the reaction. The solution was centrifuged and the supernatant fraction was read at 405 nm. The enzyme activity was calculated as nanomoles of p-nitroaniline formed per minute per milligram protein using a molar extinction coefficient of p-nitroaniline of 1.74 × 103 M−1 cm−1.
Nitrite determination
The amount of NO released by GTN was measured as its stable oxidative metabolite, nitrite (NO2 −), as described earlier. 14 Briefly, 1.0 mL of PMS (10%) was incubated at 37°C with 3 and 6 mg of GTN for 30 min. PMS was mixed with an equal volume of Griess reagent (one part 0.1% naphthyl ethylenediamine dihydrochloride in H2O and one part 1.32% sulfanilamide in 60% acetic acid) and allowed to stand at room temperature for 15 min. The absorbance at 540 nm was measured and the nitrite concentration was determined using sodium nitrite as a standard.
Estimation of serum transaminases activity
Serum transaminases activity was determined by the method of Reitman and Frankel. 33 First, 0.5 mL of α-ketoglutarate (2 mM) was incubated for 5 min at 37°C in a water bath. Then, serum (0.1 mL) was added, and the volume was adjusted to 1.0 mL with sodium phosphate buffer. The reaction mixture was incubated at 37°C for 20 min. The reaction was terminated with an equal volume of TCA (10% w/v), stored in ice, and then centrifuged at 4000 × g for 5 min. To the supernatant, 0.5 mL of 2,4 dinitrophenylhydrazine (1.0 mM) was added. The reaction mixture was left for another 30 min at room temperature. Finally, color was developed with the addition of 5 mL of sodium hydroxide (0.4 N) at 505 nm. 33
Estimation of LDH activity
LDH activity was determined by the method of Wroblewski and Ladue. 34 The reaction mixture containing 2.7 mL of phosphate buffer (0.1 mL, pH 7.4), 0.01 mL serum, and 0.1 mL of reduced nicotinamide adenine dinucleotide was incubated for 20 min. To this, 0.1 mL of pyruvate (2.5 mg mL−1) was added. The absorbance was read for 5 min at an interval of 30 s. The activity was expressed as milli international units (IU). 34
Histopathological studies
A few millimeter-thick midsections of livers from each group were excised and processed for light microscopy to validate biochemical findings. The histopathological process starts with fixing of liver tissue specimens in 10% neutral-buffered formalin solution, then the blocks in paraffin for microtome sections were prepared (5–6 µm thick) and stained with hematoxylin and eosin (H&E). The segments of liver were examined under high-resolution light microscope with photographic facilities by pathologist who was uninformed of sample assignment to experimental groups.
Estimation of protein
Protein in all samples was determined by the method of Lowry et al using bovine serum albumin as a standard. 13,35
Statistical analysis
Values were expressed as means ± SEM. The level of significance between different groups is based on Dunnett’s t test, followed by analysis of variance (ANOVA). One-way ANOVA was used to calculate the statistical significance between various groups. The value of p < 0.05 was considered to be statistically significant.
Results
Lipid peroxidation
The effect of GTN administration on CCl4-induced LPO is shown in Figure 1. CCI4 administration to rats resulted in a significant (p < 0.001) increase in LPO by 300%. GTN administration to CCl4-intoxicated rats resulted in the inhibition of LPO in a dose-dependent manner. Further increase in LPO following administration of NO inhibitor,
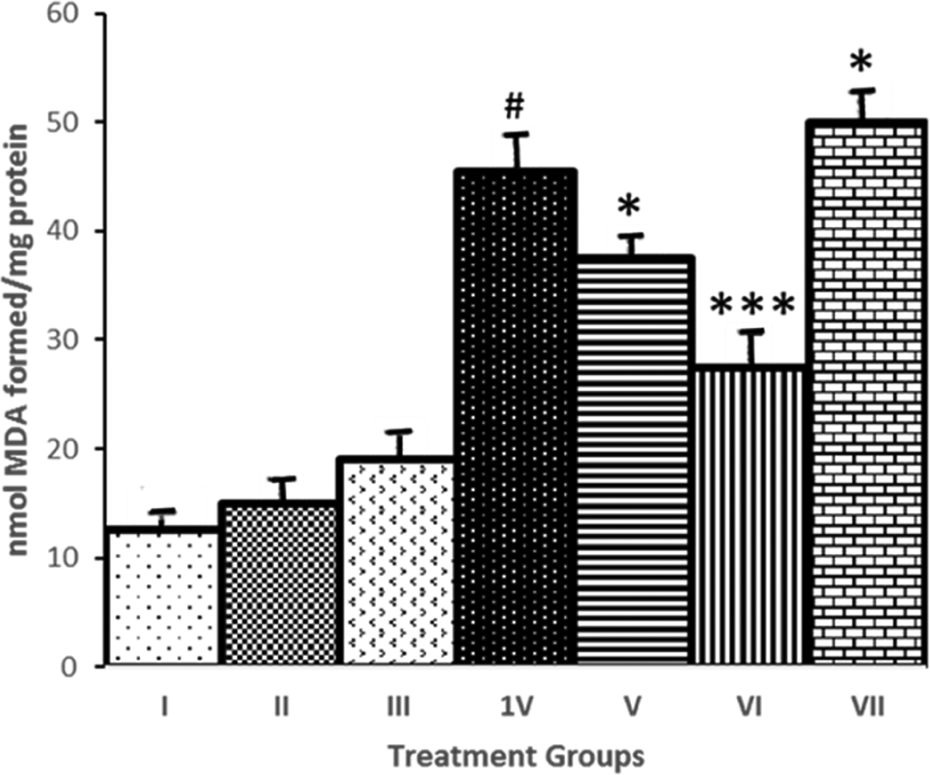
Modulatory effect of GTN on CCl4-induced hepatic microsomal LPO. Saline, (II) GTN, (III)
GGT activity
The effect of GTN administration on CCl4-induced modulation of GGT enzyme activity is shown in Figure 2. CCI4 administration resulted in a significant (p < 0.001) elevation in GGT activities (47% and 160%), when compared to saline-treated control. Administration of GTN at doses 3 and 6 mg kg−1 b. wt. to CCl4-treated animals resulted in a dose-dependent alleviation of GGT activity. A dose-dependent amelioration with 101% decrease in 6 mg kg−1 b. wt in the activity of GGT was observed at higher dose of GTN.

Modulatory effect of GTN on CCl4-induced hepatic GGT activity. (I) saline, (II) GTN, (III)

Modulatory effect of GTN on CCl4-induced histopathological changes (H&E staining). (a) saline, (b) GTN, (c)
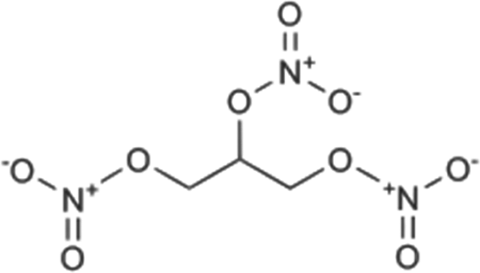
Structure of GTN. GTN: glyceryl trinitrate.
Serum transaminases (ALT and AST) and LDH activity
The effect of GTN administration on CCl4-induced release of hepatic enzymes AST, ALT, and LDH in serum is shown in Table 1. Administration of CCl4 resulted in the significant (p < 0.001) elevation of hepatic enzymes AST, ALT, and LDH in serum by 2.65-, 3.18-, and 1.68-folds increase in their respective control. Administration of GTN subsequent to CCl4-treated animals significantly (p < 0.001) attenuated the elevated levels of AST and ALT enzymes with concomitant inhibition of LDH activity at significance (p < 0.01) in a dose-dependent manner. Moreover,
Modulatory effect of GTN on CCl4-induced levels of serum transaminases (ALT and AST), and Lactate dehydrogenase (LDH).a

AST: aspartate transaminase; ALT: alanine transaminase; LDH: lactate dehydrogenase; GTN: glyceryl trinitrate;
aD1: rats were administered CCl4 (1 mL kg−1 body weight; i.p.) followed by GTN (3 mg kg−1 body weight; i.p.) after 1 h of CCl4 administration; D2: Rats were administered CCl4 (1 mL kg−1 body weight; i.p.) followed by GTN (6 mg kg−1 body weight) after 1 h of CCl4 administration. Data represent mean ± SE of six animals. Dose regimen and treatment protocol are described in the text.
b p < 0.001: saline-treated group served as control for AST; aspartate group II, III and IV.
c p < 0.01: saline-treated group served as control for AST; aspartate group II, III and IV.
d p < 0.001: CCl4-treated group served as control for groups V, VI and VII.
e p < 0.01: CCl4-treated group served as control for groups V, VI and VII.
Antioxidants activity
The effect of GTN administration on CCl4-mediated depletion in hepatic antioxidants is shown in Table 2.
Modulatory effect of GTN on CCl4-induced levels of hepatic glutathione-metabolizing enzymes.a
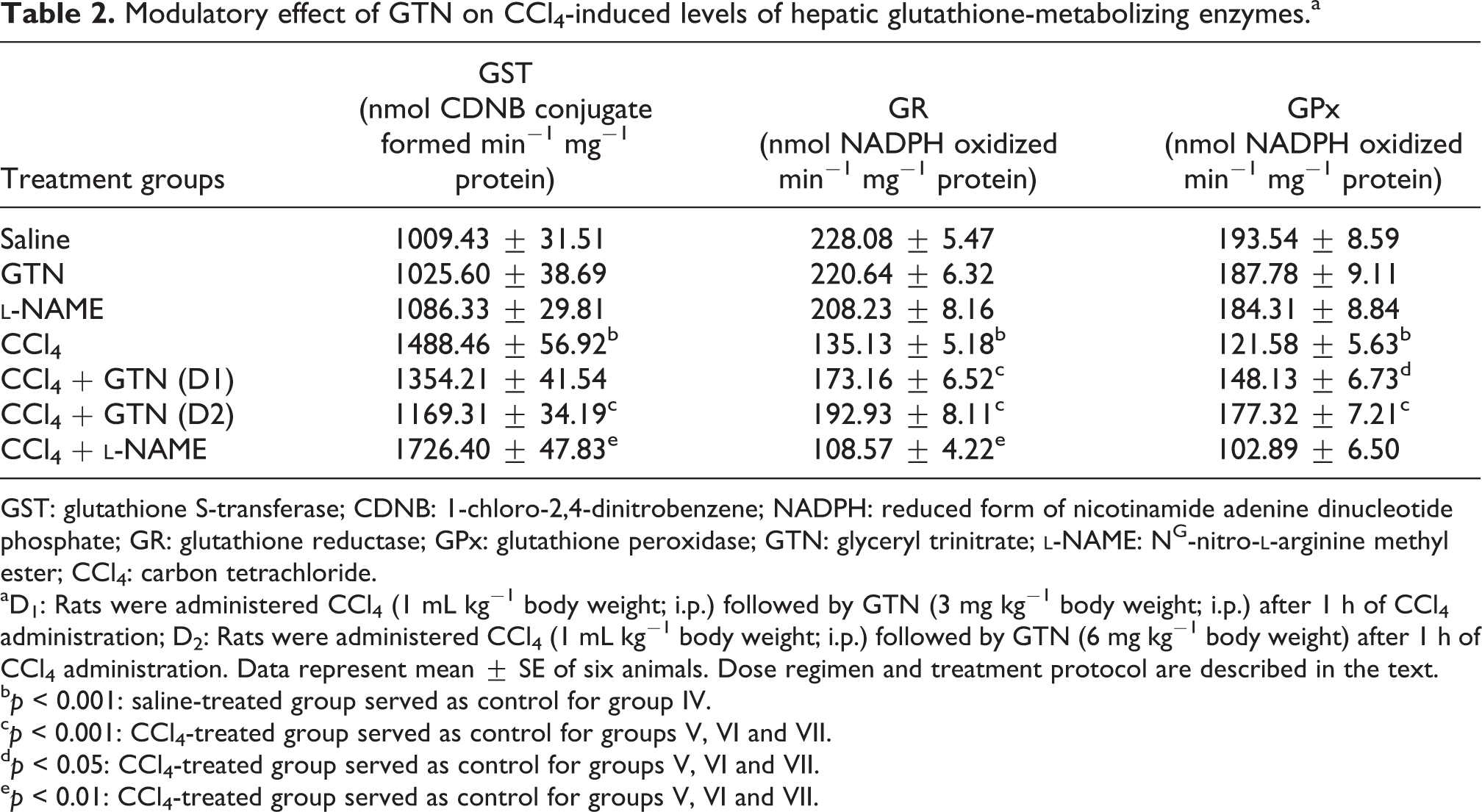
GST: glutathione S-transferase; CDNB: 1-chloro-2,4-dinitrobenzene; NADPH: reduced form of nicotinamide adenine dinucleotide phosphate; GR: glutathione reductase; GPx: glutathione peroxidase; GTN: glyceryl trinitrate;
aD1: Rats were administered CCl4 (1 mL kg−1 body weight; i.p.) followed by GTN (3 mg kg−1 body weight; i.p.) after 1 h of CCl4 administration; D2: Rats were administered CCl4 (1 mL kg−1 body weight; i.p.) followed by GTN (6 mg kg−1 body weight) after 1 h of CCl4 administration. Data represent mean ± SE of six animals. Dose regimen and treatment protocol are described in the text.
b p < 0.001: saline-treated group served as control for group IV.
c p < 0.001: CCl4-treated group served as control for groups V, VI and VII.
d p < 0.05: CCl4-treated group served as control for groups V, VI and VII.
e p < 0.01: CCl4-treated group served as control for groups V, VI and VII.
CCI4 administration resulted in a significant (p < 0.001) elevation in GST (47%) with concomitant decrease in the activities of GR and GPx (41% and 37%), when compared to saline-treated control. Administration of GTN at doses 3 and 6 mg kg−1 b. wt. to CCl4-treated animals resulted in a dose-dependent alleviation of GST activity. A 31% decrease in the activity of GST was observed at higher dose of GTN. Similarly, a dose-dependent amelioration in the activities of GR and GPx by 26% and 28% was observed in these animals.
Amount of NO generated by GTN
The maximum amount of nitrite detected following incubation of GTN with PMS for a total of 45 min yielded 0.8 ± 0.2 and 1.5 ± 0.3 mM/g tissues, at 3 and 6 mg doses of GTN.
Histopathological studies
The effect of GTN administration on CCl4-induced changes in liver histology is shown in Figure 3. It was observed that CCI4 administration to rats caused necrosis, fatty degeneration, sinusoidal dilatation, blood vessel congestion, and derangement of hepatocytes. However, these changes were ameliorated by GTN treatment in a dose-dependent manner as shown in Figure 3. However, GTN and
Discussion
The role of NO in hepatic disease is complex. Previously it has been shown that NO can protect the liver by scavenging lipid radicals and inhibiting the LPO chain reaction. 36 Alternatively, hydroxyl radicals formed by the reaction of NO and superoxide anion via peroxynitrite may result in hepatotoxicity. 37 In this study, we sought to characterize the role of NO donor and inhibitor in CCl4-induced hepatotoxicity.
CCl4 is a well-known hepatotoxic agent that induces liver injury, which is regarded as the analog of liver damage caused by a hepatotoxins in man.
38
It is characterized by expression of pro-inflammatory mediators with evidence of hepatocellular necrosis.
5
CCl4-induced hepatotoxicity depends on the reductive dehalogenation of CCl4 catalyzed by cytochrome P450 in the liver cell endoplasmic reticulum leading to the generation of an unstable complex of trichloromethyl radical. Covalent binding of the trichloromethyl radical to cell protein is considered the initial step in a chain of events that eventually leads to LPO of the cell membrane in addition to inducing cell membrane dysfunction leading to hepatocellular damage.
39
Although GTN administration dose dependently decreased the fatty changes and vacuolation, yet the degree of decrease in the fatty changes and vacuolation was clear at higher dose of GTN. However,
Cellular thiol status is an important consideration in hepatotoxicity and oxidative stress. 44 It has been speculated that NO may protect against oxidative injury by modulating intracellular GSH levels. 45 GST catalyzes electrophilic conjugation of reactive intermediates with GSH, thus, enhancing their elimination as water-soluble products. GPx has been shown to regulate H2O2, lipid hydro-peroxide, and OH radical levels in cytosol and mitochondria, 46 while GR catalyzes recovery of GSH from glutathione disulfide utilizing NADPH. 47 GTN-mediated release of NO reportedly inhibits cytochrome P450 enzyme by binding to its P450 heme moiety. 48,49 Since, CCI4 elicits its toxic potential through cytochrome P450-dependent activation, it could be suggested that GTN-mediated NO delivery in CCl4-intoxicated rats might have inhibited the metabolic activation of CCl4, thereby, reducing production of CCl4 metabolites and dependent oxidative stress in liver thus reducing the hepatic injury. GTN administration caused a significant decrease in GST activity with concomitant recovery of GR and GPx activities in a dose-dependent manner. A plausible explanation for this protective action might be the activation of components in the glutathione recycling pathway including GR and GPx, and intracellular glutathione content by scavenging the electrophilic metabolites of CCl4 and rendering them excretable as less toxic complex. Evidence that inhibition of GR leads to marked decrease in NO production with concomitant depletion of GSH content suggest NO as the constitutive aspect for GSH status. 50
NO plays an important role in the liver with the potential for both protection of the liver cells from injury as well as exacerbation of injury. NO could play an important role in liver injury, as it is formed by hepatocytes, Kupffer’s cells, and endothelial cells with adequate stimulus. 36 Therefore, the function of NO in liver injury is complex and paradoxical because both prooxidant and antioxidant properties have been reported. Although NO in very high concentrations induces cell injury under some conditions, the preponderance of evidence would suggest that under most conditions endogenous NO exerts protective effects in the liver. 51 There is a possibility of decrease in NO production after CCl4 administration may be due to its consumption in terminating LPO and depletion of antioxidant enzymes, caused by interaction with CCl4.
In summary, our results suggest that exogenous NO could be used to protect against CCl4-induced liver injury and oxidative stress, which greatly depend on its amount, location, and duration of generation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.