
Abstract
Periodontitis is a common infectious disease associated with destruction of periodontal ligaments and alveolar bones. CD4+ T cell-mediated immune response is involved in the progression of periodontitis. Tobacco consumption increases the risk of periodontal disease. However, the impact of nicotine on the interaction between human periodontal ligament (PDL) cells and CD4+ T cells remains unrevealed. Our study aims to investigate the effect of nicotine on PDL cells and the cocultured CD4+ T cells. The PDL cell cultures were established by explants from healthy individuals, exposed to nicotine or α-bungarotoxin (α-BTX), and incubated solely or in combination with CD4+ T cells. Afterwards, cell viability, secreted cytokines, and matrix metalloproteinases (MMPs) were evaluated. In monoculture of PDL cells, nicotine dramatically repressed cell viability and increased apoptosis. Meanwhile, α-BTX largely reversed the nicotine-induced apoptosis and increased viability of PDL cells. Compared with the monoculture, MMP-1, MMP-3, interleukin (IL)-1β, IL-6, IL-17, and IL-21 in supernatant of cocultures were markedly elevated after treatment with nicotine. Moreover, α-BTX significantly attenuated nicotine-triggered production of these components either in mono- or co-cultures. In addition, PDL cell-derived CXCL12 following nicotine treatment recruited CD4+ T cells. Above all, nicotine deteriorated periodontitis partially by promoting PDL cell–CD4+ T cell-mediated inflammatory response and matrix degradation.
Introduction
Periodontitis ranks among the most common chronic infectious diseases with a high prevalence of 5–15% in adults globally. The initiation of periodontitis is often caused by inappropriate and excessive immune response to bacteria in subgingival dental plaques, which in turn leads to loss of connective tissue attachment, abnormal alveolar bone resorption, and even tooth loss. 1 Periodontal ligament cells reside in the periodontal ligament space and play an important role in the formation of periodontal ligament via secreting extracellular matrix components such as collagen and mineralized tissue. 2 Simultaneously, PDL cells are responsible for generation of various pro-inflammatory cytokines and chemokines as well as matrix metalloproteinases in pathological events, thereby implicating in the progression of periodontal diseases. 3
Epidemiological and clinical data have demonstrated that smoking is one of the important predictors and risk factors of periodontitis and by-products deriving from tobacco oxidation have adverse impacts on clinical characteristics and progression of periodontal diseases. 4 Cigarette consumption has been shown to induce alterations in microbial profile and host response including neutrophil/monocyte activities, vascular function, and cytokine and inflammatory factor secretion, eventually leading to impairment of the reparative and regenerative activities of the periodontium. 5 As a well-known active substance in cigarette smoke, nicotine is deleterious to human gingival fibroblasts and periodontal ligament cells that account for maintaining the integrity of periodontal tissue. 6 Moreover, daily injection of nicotine exacerbates alveolar bone damage. 7 Previous study proved that α7 nicotinic acetylcholine receptor (α7 nAChR) was functionally expressed in human PDL cells and rat periodontal tissues, which was a ligand-gated ion channel and could be activated and induced by nicotine whereas partially inhibited by α-bungarotoxin (α-BTX).
Multiple lines of evidence revealed that patients with chronic periodontitis presented with higher frequency of T lymphocytes and CD4+CD25+ T cells in the inflammatory infiltrates of periodontal and gingival tissues, implying that CD4+ T cell-mediated immune response might be involved in the immunopathology of periodontitis. 8,9 Furthermore, emerging research studies have shown that IL-17 and Th17 cells (IL-17 producing CD4+ T cells) are presented in human periodontitis lesions and may mediate progression of the inflammatory disease. However, effects of nicotine on inflammatory response and extracellular matrix remain unrevealed. In the current study, we will examine the effects of nicotine on PDL cells’ viability and apoptosis. Subsequently, we will focus on the effects of nicotine on the production of inflammatory factors and chemokines as well matrix metalloproteinases in PDL cells and the cocultures with CD4+ T cells.
Materials and methods
Cell culture
Primary culture of fibroblasts was established by explant culture from periodontal ligaments of three young healthy individuals who underwent orthodontic surgeries. The explants, obtained from the root of premolar teeth, were grown in RPMI-1640 medium containing 15% fetal calf serum, growth factors (basic fibroblast growth factor and insulin), and antibiotics (penicillin (50 U/ml) and streptomycin (50 mg/ml)) at 37°C in humidified air with 5% carbon dioxide (CO2). Cells from passages four to six were used for all experiments. This study complied with Declaration of Helsinki and was approved by the ethics committee at School of Stomatology, The Fourth Military Medical University, China.
Purification of CD4+ T cell
Peripheral blood mononuclear cells, collected from healthy nonsmoking volunteers, were isolated using density gradient centrifugation (Ficoll-Hypaque solution, Sigma; St Louis, Missouri, USA). Afterwards, CD4+ T cells were purified by immunomagnetic depletion using the human CD4+ T cell isolation kit II according to the manufacture’s protocol (Miltenyi Biotec, Rostock, Germany). Subsequently, the purified CD4+ T lymphocytes were cultured in RPMI-1640 medium supplemented with 15% fetal calf serum and antibiotics (penicillin (50 U/ml) and streptomycin (50 mg/ml)). To examine the effect of nicotine and α-BTX on the interaction particularly between PDL cells and CD4+ T cells, monoculture of PDL cells and coculture of PDL cells and CD4+ T cells (1:4) were established. A semipermeable membrane (0.4 µm; Millipore, Germany) was used to construct the coculture system. The PDL cells were pretreated with nicotine (10−5 M), α-BTX (10−8 M), and nicotine combined with α-BTX for 24 h at 37°C in humidified air with 5% CO2, respectively. Then the monoculture and coculture systems were established and cells were cultured for 48 h.
Cell viability assay
Viability of PDL cells that were treated with nicotine, α-BTX, or their combination was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In brief, cells were seeded in 96-well plates at a density of 1 × 104 cells per well in 300 µL medium in the presence of nicotine and α-BTX as well as nicotine combined with α-BTX, and the cells were then incubated at 37°C for 24 h. After treatment, 50 µL MTT solutions (Sigma-Aldrich, St Louis, Missouri, USA) dissolved in culture medium at a final concentration of 0.5 mmol/l were added to each well and the plates were incubated at 37°C for another 4 h. Afterwards, the culture medium was discarded and 300 μL dimethyl sulfoxide (Sigma-Aldrich, ST, USA) was then added to each well, shaking for 20 min to solubilize MTT tetrazolium crystal. Finally, the absorbance was measured at 570 nm using a Benchmark Plus microplate reader (Bio-Ran; Hercules, California, USA).
Apoptosis
The treated PDL cells (1–5 × 105/well) were seeded and incubated overnight and were then digested with trypsin before being collected. Then, Vybrant® apoptosis assay kit (Invitrogen, Carlsbad, California, USA) was used to assess the apoptotic cells using annexin V/propidium iodide (PI) double staining. The stained cells were analyzed by flow cytometry.
Western blotting
Expressions of TIMP-1 and β-actin in monoculture and coculture were measured by Western blotting using commercially available antibodies obtained from Abcam (Cambridge, Massachusetts, USA). The whole cell lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and subsequently transferred to a polyvinylidene fluoride membrane. Blots were visualized using Amersham Western blot detection reagent (GE Healthcare; Piscataway, New Jersey, USA).
Measurements of cytokine production using enzyme-linked immunosorbent assay assay
To evaluate the concentrations of IL-1, IL-6, IL-17, IL-21, and CXCL12 in mono- and co-culture medium in response to various treatments, commercially available enzyme-linked immunosorbent assay (ELISA) kits (Hoelzel Diagnostika, Cologne, Germany) were used and all the experiments were performed according to the manufacturer’s instructions.
Transwell assay
For the chemotaxis assay of CD4+ T cell, transwell cell culture inserts (3-μm pore; BD Biosciences, Belgium) were placed in conditioned medium of PDL cells pretreated with nicotine, α-BTX, or their combination in the lower compartment. Afterwards, 3 × 105 CD4+ T cells were seeded on the insert and allowed to migrate overnight at 37°C. The cells that migrated into the lower well were stained using Diff-Quick kit (Dade Behring, Deerfield, Illinois, USA). Pictures were taken with a microscope and migrated cells were counted with Image J software 1.38e (National Institutes of Health, MD, USA) using eight randomly chosen fields.
Statistical analysis
Data are expressed as means ± SD. All statistical analyses were performed using SPSS 13.0 (SPSS; Chicago, Illinois, USA). Statistical comparisons were performed by one-way analysis of variance followed by the Student-Newman-Keuls test. A p < 0.05 was considered statistically significant.
Results
Effects of nicotine and α-BTX on PDL cell viability
The direct effects of nicotine and α-BTX as well as nicotine in combination with α-BTX on the growth of PDL cells were determined using MTT assays over 4 days. Compared to the control, PDL cells significantly decreased following the treatment with 10−5 M nicotine at 48 h (p < 0.05), while the inhibition of α7 nAChR with its antagonist α-BTX didn’t significantly change the cell viability within 96 h after the incubation. Moreover, in comparison with the PDL cells exposed to nicotine, the administration of α-BTX dramatically rescued the diminished cell viability induced by nicotine at 48 h (p < 0.05; Figure 1).
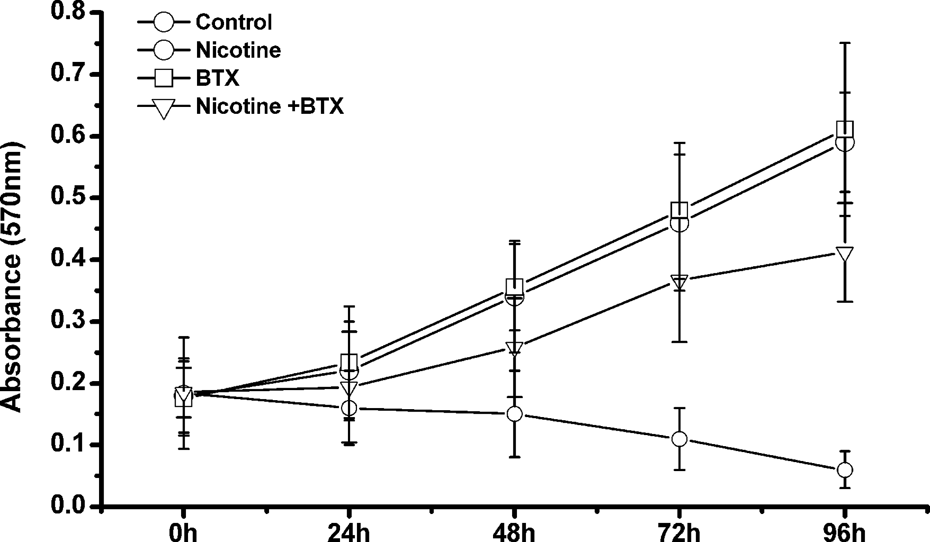
Impacts of nicotine and α-BTX on PDL cell viability. PDL cells were pretreated with nicotine, α-BTX and nicotine combined with α-BTX. MTT assays were conducted to analyze PDL cell viability in response to nicotine and/or α-BTX treatments at 24, 48, 72, and 96 h. α-BTX: α-bungarotoxin; PDL: periodontal ligament; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Effects of nicotine and α-BTX on PDL cell apoptosis
PDL cell apoptosis was detected using PI/annexin V-fluorescein isothiocyanate via flow cytometry, as shown in Figure 2(a) and (b), after incubation with nicotine for 48 h, the apoptotic cells markedly increased to 17.62 ± 3.44% compared to the control of 2.12 ± 1.01% (p < 0.05). Simultaneously, α-BTX seemed to have no obvious impact on PDL cell apoptosis compared with the control when evaluated after incubation with α-BTX for 48 h (2.26 ± 0.97% vs 2.12 ± 1.01%, p > 0.05; Figure 2(c)). However, the combination of α-BTX with nicotine significantly alleviated cell apoptosis of PDL cells compared with the application of nicotine solely (6.57 ± 2.36% vs 17.62 ± 3.44%, p < 0.05; Figure 2(d)).

Effects of nicotine and α-BTX on PDL cell apoptosis. (a) Annexin V/PI double staining assays were performed to measure apoptotic cells following the treatments with nicotine, α-BTX, and nicotine combined with α-BTX. (b) Percentage of apoptotic PDL cells in response to various treatments. *p < 0.05: compared to the control; #p < 0.05: compared to nicotine-treated cells. α-BTX: α-bungarotoxin; PDL: periodontal ligament; PI: propidium iodide.
Nicotine exacerbates extracellular matrix breakdown in PDL cells and the cocultures with CD4+ T cells
In order to investigate roles of nicotine in the homeostasis of extracellular matrix, we determined the expression of MMP-1, MMP-3, and TIMP in PDL cells as well as in the cocultures of PDL cells with CD4+ T cells using ELISA or Western blotting. It was demonstrated that after treatment with nicotine for 48 h, the secreted MMP-1 and MMP-3 significantly elevated while TIMP reduced both in PDL cells and the coculture of PDL cells with CD4+ T cells compared with their own matched controls (p < 0.05). At the same time, the coculture system exhibited higher levels of MMP-1 and MMP-3 compared to the monoculture of PDL cells (p < 0.05). In addition, the production of MMP-1 and MMP-3 as well as TIMP changed neither in PDL cells nor in the coculture of PDL cells with CD4+ T cells in response to the treatment with α-BTX (p > 0.05). Furthermore, α-BTX markedly suppressed nicotine-stimulated generation of MMP-1 and MMP-3 while promoted TIMP expression in mono- and co-culture systems, implying that α-BTX contributed to the inhibition of nicotine-induced degradation of extracellular matrix (Figure 3).

Nicotine promotes ECM degradation in PDL cells. ELISA analysis of MMP-1 (a) and MMP-3 (b) after stimulation by nicotine, α-BTX, and nicotine combined with α-BTX in PDL cells and cocultures of PDL cells with CD4+ T cells. (C) Western blotting analysis of TIMP-1 in PDL cells and cocultures of PDL cells with CD4+ T cells. *p < 0.05, **p < 0.01: compared to the control; #p < 0.05, ##p < 0.01: compared to nicotine-treated cells; &p < 0.05: compared to monocultured PDL cells. α-BTX: α-bungarotoxin; ECM: extracellular matrix; PDL: periodontal ligament; ELISA: enzyme-linked immunosorbent assay; MMP: matrix metalloproteinase.
Nicotine induces secretion of cytokines in PDL cells and the cocultures with CD4+ T cells
To evaluate the effect of nicotine on the inflammatory response in PDL cells and PDL cells cocultured with CD4+ T cells, the levels of IL-1β, IL-6, IL-17, and IL-21 in the supernatants of each culture system were identified using ELISA assays. As shown in Figure 4(a) and (b), IL-1β and IL-6 in both mono- and co-cultures significantly increased in response to stimulation with nicotine compared with their own controls (p < 0.05). Moreover, IL-1β and IL-6 in cocultures of PDL cells and CD4+ T cells were significantly higher than PDL cells sole culture (p < 0.05). α-BTX largely attenuated the nicotine-elicited upregulation of IL-1β and IL-6 both in PDL cells and cocultures with CD4+ T cells. Compared to the control, IL-17 and IL-21 had no significant changes in monoculture of PDL cell medium following the treatment with nicotine. However, they displayed statistically higher levels in the supernatant of cocultures after stimulation with nicotine than the control in mono and coculture (p < 0.05). Likewise, α-BTX restrained the generation of IL-17 and IL-21 induced by nicotine in the coculture system (Figure 4(c) and (d)).

Nicotine increases generation of inflammatory cytokines in PDL cells and cocultures of PDL cells with CD4+ T cells. ELISA approaches were used to evaluate the concentration of IL-1 (a), IL-6 (b), IL-17 (c), and IL-21 (d) following stimulation with nicotine and/or α-BTX in PDL cells and PDL cells cocultured with CD4+ T cells. *p < 0.05, **p < 0.01: compared to the control; #p < 0.05: compared to nicotine-treated cells; &p < 0.05: compared to monocultured PDL cells. PDL: periodontal ligament; ELISA: enzyme-linked immunosorbent assay; α-BTX: α-bungarotoxin; ELISA: enzyme-linked immunosorbent assay; IL: interleukin.
Nicotine-induced CXCL12 in PDL cells triggers the migration of CD4+ T cells
Emerging evidence revealed that CD4+ T cells infiltrated into periodontal and gingival tissues and participated in the progression of periodontitis, we therefore sought to examine whether nicotine played a role in recruiting CD4+ T cells and the underlying mechanism. Using a transwell assay, we found that the migrated CD4+ T cells markedly increased in the conditioned medium of PDL cells that were pretreated with nicotine in comparison to the control. While, addition of α-BTX-incubated conditioned medium of PDL cells didn’t significantly change the amounts of migrated CD4+ T cells. However, the inhibitory effect of α-BTX was noticeable in the migration assay following nicotine stimulation (Figure 5(a)). Simultaneously, we measured the levels of CXCL12 in the medium of PDL cells that were pretreated with nicotine and α-BTX solely or combined with nicotine. It was demonstrated that nicotine dramatically induced the upregulation of CXCL12 compared to control, which was significantly reduced by α-BTX compared to the nicotine-treated cells (p < 0.05; Figure 5(b)).

Nicotine-induced secretion of CXCL12 in PDL cells recruits CD4+ T cells. (a) CD4+ T cells were incubated in the conditioned medium of PDL cells that were pretreated with nicotine, α-BTX, and nicotine combined with α-BTX, then the migrated cells were determined using transwell assays. (b) ELISA analysis of the concentration of CXCL12 in PDL cells in response to the treatments with nicotine, α-BTX, and nicotine combined with α-BTX. All experiments were performed in triplicate. *p < 0.05: compared to the control; #p < 0.05: compared to nicotine treated cells. α-BTX: α-bungarotoxin; PDL: periodontal ligament; ELISA: enzyme-linked immunosorbent assay.
Discussion
Periodontitis is a periodontopathogen-elicited infectious disease that causes chronic inflammatory and immune response against periodontal structures. Nicotine has been documented as an independent risk factor of the initiation and progression of periodontal diseases. However, the underlying mechanisms regarding the role of nicotine in periodontitis remain unknown. In the current study, we examined the effects of nicotine on PDL cell behaviors and the cross talk between PDL cells and CD4+ T cells concerning the production of extracellular proteins and cytokines.
Tobacco consumption exacerbates periodontal tissue destruction, pocket formation, and alveolar bone resorption in periodontitis. 10 As a major detrimental component of tobacco, nicotine contributes to the induction of α7 nAChR in human PDL cells and rat periodontal tissues. 11 In this study, we also found that inhibition of α7 nAChR using its specific antagonist α-BTX apparently attenuated nicotine-induced cytotoxicity in PDL cells. Moreover, incubation with nicotine significantly repressed cell viability, which was in accordance with the previous data reported by James et al. 12 Likewise, nicotine application promoted PDL cell apoptosis, which was associated with the collapse of periodontal tissues. Therefore, the influence of nicotine on PDL cell behavior may facilitate the onset of periodontal diseases and α7 nAChR blockade using α-BTX will alleviate the impaired cell function and viability, which will be a new strategy for the treatment of periodontitis.
Growing evidence has clarified that CD4+ T cell-mediated inflammatory response is associated with the onset of periodontitis. CD4+ T cells are presented in Porphyromonas gingivalis-induced chronic periodontitis tissues. 13 Meanwhile, PDL cells might be implicated in inflammatory mechanisms and transition to an adaptive immune response in periodontal tissues. 14 In the current study, we found that coculture of PDL cells with CD4+ T cells exhibited higher levels of cell cytokines such as IL-6, IL-1β, IL-17, and IL-21 after nicotine stimulation while IL-17 and IL-21 were almost negligible in the sole culture of PDL cell incubated with nicotine. Based on the above observations, we concluded that nicotine might elicit the generation of IL-6 and IL-1β both in PDL cells and CD4+ T cells. However, the cross talk between PDL cells and CD4+ T cells regarding the secretion of IL-6 and IL-1β in response to nicotine exposure was unraveled.
Th17 cells are identified as an independent lineage of CD4+ T cells that secrete a distinctive set of immunoregulatory cytokines, including IL-17A, IL-17F, IL-22, and IL-21. These cytokines collectively play roles in inflammation and autoimmunity and in the response to extracellular pathogens. In addition, a more recent study has demonstrated that the application of IL-17 is responsible for the upregulation of MMP-1 and MMP-13 and downregulation of MMP-2, MMP-9, and TIMP-1 in human periodontal ligament cells. 15 Therefore, we conclude that multiple signaling pathways are implicated in nicotine-induced matrix breakdown. Emerging data have revealed that Th17 cells are presented in chronic lesions of human periodontal disease, and elevated IL-17, TGF-β, IL-1β, IL-6, and IL-23 are observed in gingiva from patients with periodontitis. Meanwhile, IL-17 and the bone resorption factor RANKL are dramatically elevated in the alveolar bone of diseased patients. 16,17 It was also claimed that Th17 cells developed in response to TGF-β, IL-1β, and IL-6 signaling. 18,19 Therefore, in the current study nicotine-induced generation of IL-1β and IL-6 in PDL cells and CD4+ cells may further promote the development of Th17 cells.
Above all, nicotine inhibited cell viability, promoted apoptosis, and elicited the production of inflammatory cytokines, chemokines, and MMPs in PDL cells, which were more severe in the cocultures with CD4+ T cells. However, inhibition of α7 nAChR using α-BTX largely attenuated these effects induced by nicotine. Therefore, nicotine exacerbated inflammatory response, matrix degradation, and recruitment of leukocytes, which were associated with the onset and progression of periodontal diseases. Further studies addressing the cross talk between CD4+ T cells and PDL cells during inflammatory responses in periodontitis are needed.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (NSFC grant no. 81200788).