
Abstract
Stem cell transplantation has indicated great promise for cell therapy in a wide range of diseases, but poor and insufficient viability of cells within damaged tissues has limited its potential therapeutic effects. 17 β-Estradiol (E2) is a steroid hormone that plays an important role in expression of many genes and regulating proliferation, viability, and intracellular redox status in different cell types. In this study, we aimed to assess the effect of E2 on bone marrow-derived mesenchymal stem cells (BM-MSCs). Apoptosis was induced by serum deprivation (SD), and cells were exposed to E2 in the presence or absence of serum for varying periods of time, after which cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Expression of proapoptotic and antiapoptotic proteins after exposure to E2 was examined by Western blotting. The ability of E2 to prevent reactive oxygen species (ROS) production was also measured. The results indicated that E2 significantly enhanced the viability of the cells and protected BM-MSCs against SD-induced overproduction of ROS. It could reduce lipid peroxidation, total antioxidant power, and also Bax/Bcl-2 ratio as well as expression of caspase-3. Taken together, our data support that E2 treatment protects BM-MSCs against SD-induced damage by regulating ROS production and upregulation of antiapoptotic/proapoptotic proteins ratio.
Introduction
Emerging evidences show that bone marrow-derived mesenchymal stem cells (BM-MSCs) play a crucial role in cell therapy. 1 High expansion capability, ease of isolation, trophic action, and ability to move toward injured sites have made BM-MSCs an attractive candidate for regenerative therapy. 2,3 Moreover, immunomodulatory properties of MSCs also make them a suitable treatment choice for a wide range of diseases. 4 However, a major obstacle in cell-based therapies is inefficient viability of transplanted stem cells in injured tissues. Previous studies have demonstrated that a large part of transplanted stem cells do not survive well within targeted tissues 5,6 and most of them are eliminated by the immune system and die by apoptosis. 5,7 Thus, enhancing the viability of MSCs at sites of injury may improve the overall therapeutic efficacy of BM-MSCs’ transplantation.
It was demonstrated that cultured cells undergo apoptosis in response to ischemia components such as hypoxia or serum deprivation (SD) and nutrient deprivation.
7
–9
SD has been widely accepted as an apoptotic model that mimics ischemic components in vivo.
10,11
Similar to many kinds of cells, BM-MSCs are also dependent on serum for their survival, so that serum withdrawal causes cell death of apoptotic type.
8
SD could generate reactive oxygen species (ROS; including superoxide anion
Natural estrogens showed a significantly higher level of antioxidant activity than vitamins E and C. 16,17 Among them, 17β estradiol (E2) has multifunctional roles that influence survival, growth, and differentiation of cells in several tissues. 18,19 Antiapoptotic effect of E2 has also been demonstrated in different cell types. E2 inhibits oxidative stress-induced apoptosis by enhancing Bcl-2 expression and phosphorylating cyclic adenosine monophosphate/protein kinase A pathway in keratinocytes. 20 Estrogen also increased the survival of monoblastoid cells by preventing apoptosis induced by tumor necrosis factor α. 21 E2 also protects MSCs against H2O2-induced apoptosis and can reduce the damage of donor stem cells. 22 Moreover, E2 has indicated to protect human adipose stem cells against cytotoxicity and to elevate survival of transplanted cells in spinal cord injury. 23
Therefore, in this study, we tried to examine the protective effects of E2 on SD-induced oxidative stress and apoptosis in BM-MSCs.
Materials and methods
Rat BM-MSCs’ isolation and cell culture
BM-MSCs were isolated from the bilateral femurs and tibias of the male Wistar rats (8 weeks old, 150–200 g) by removing the epiphyses and flushing the shaft with minimal essential medium α (MEM-α; Gibco, Invitrogen, Carlsbad, California, USA). The cells were grown in MEM-α supplemented with 10% fetal bovine serum (FBS; Gibco, Invitrogen) and 1% penicillin–streptomycin (Gibco, Invitrogen) in a cabon dioxide (CO2)-humidified chamber of 95% air and 5% CO2. After 48 h, nonadherent cells were removed, and culture medium was changed every 3 days. Cells at passage 2–4 were used to perform the experiments. E2 (CAS RN 4956-37-0, Sigma Aldrich, St Louis, Missouri, USA) was dissolved in ethanol (99.5%) and stock solution stored at 4°C. BM-MSCs were exposed to 15% serum (control), serum-free medium, and serum-free medium + E2 (1–1000 nM) for 24–48 h. This study was approved by Ethical Committee of Iran University of Medical Sciences.
BM-MSC characterization
Cell surface markers of BM-MSCs were quantified by fluorescence-activated cell sorting (FACS) analysis of a homogenous population of fibroblast-like cells obtained from passages 2–4. For direct labeling, a total of 2 × 105 cells were incubated with fluorescein isothiocyanate-conjugated monoclonal rat antimouse antibodies positive to cluster of differentiation 44 (CD44), CD90 (BD Biosciences Pharmingen, California, USA), CD73, CD105 (Santa Cruz Biotechnology, California, USA), and CD106 (Biolegend, San Diego, California, USA) and negative to CD31, CD45 (BD Biosciences Pharmingen) and CD34 (Santa Cruz Biotechnology) for 40 min on ice in darkness. Unstained cells were used as negative control. The analysis was then carried out by using a FACSCalibur cytometer (Becton and Dickinson Co., Franklin Lakes, New Jersey, USA) and CellQuest software (Becton and Dickinson Co.).
MTT reduction assay
This colorimetric assay is an established method of determining viable cell number in cytotoxicity and proliferation studies. Reduction of tetrazolium salts by the mitochondrial succinate dehydrogenase to a blue formazan product is directly proportional to the number of living cells. Briefly, BM-MSCs were seeded in 96-well plates at a density of 5000 cells/well and then the cells were exposed to different concentrations of E2 (1–1000 nM) in the presence or absence of FBS for 24 and 48 h. Nontreated BM-MSCs were used as control (without FBS for negative and with 15% FBS for positive control). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma Aldrich) was added to each well and incubated for 2 h at 37°C in darkness. Medium was then discarded, and 100 μl ofdimethyl sulfoxide (Sigma-Aldrich) was added. At 570 nm, absorbance was measured by a microplate reader (BioTek ELX800, Winooski, Vermont, USA).
Measurement of cell apoptosis by PI
Propidium iodide (PI) was used to assess the percentage of nonviable cells. After conditioning with E2, treated and control cells were washed with phosphate-buffered saline (PBS) and trypsinized. Then, cells were centrifuged for 10 min at 1500 r/min. The pellets were resuspended in PBS, and the cells were fixed with ice-cold 70% ethanol for 24 h at 4°C. The pellets were suspended in PBS-containing 10 mg/ml RNAase A and PI (50 μg/ml) for staining. After 30 min incubation at 37°C in the dark, the PI-DNA complex was measured using FACS Diva (Becton and Dickinson Co.). Subsequent analysis to determine the percentage of cells in various stages of cell cycle was done using FlowJo (Tree Star; Ashland, OR, USA).
Measurement of intracellular ROS
2′-7′-Dichlorodihydrofluorescein diacetate (DCF-DA) is a lipophilic and nonfluorescent compound that is widely used to evaluate ROS production in cultured tissues. After passing through the plasma membrane, it was oxidized to the fluorescent DCF by a process usually considered to involve ROS. In this assay, cells were seeded in 24-well plates and exposed to serum-free medium with increasing concentrations of E2 for 24 and 48 h. After incubation periods, the medium was removed, and the cells were rinsed and incubated with DCF-DA for 30 min. ROS production was measured using 485/20 nm excitation and 528/20 nm emission wavelengths by a fluorescence plate reader.
Protein extraction and Western blot analysis
Cells were washed twice with ice-cold PBS and lysed by radio immunoprecipitation assay lysis buffer (Roche, Applied Science, Germany) including protease inhibitors and centrifuged for 30 min at 12,000g at 4°C. Total protein concentration was measured by Bradford protein assay (Bradford 1976). Then, equal amounts of samples (50 μg) were loaded and separated by a 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel and transferred onto a polyvinylidene fluoride membrane (BioRad, Hercules, California, USA). The membranes were incubated with the following primary antibodies: mouse monoclonal antibody anti-caspase 3 (1:500; Sigma Aldrich), rabbit polyclonal antibody anti-Bax (1:200, Abcam, Cambridge, UK), mouse polyclonal anti-Bcl2 (1:1000, Abcam), and rabbit anti-β-actin (1:5000, Cell Signaling Technology, Danvers, Massachusetts, USA). Then, the membranes were incubated with secondary antibody (anti-mouse immunoglobulin G and anti-rabbit antibody conjugated with horseradish peroxidase Cell Signaling Technology, Danvers) for 1 h. The immunocomplexes were visualized using an enhanced chemiluminescence kit (Amersham Biosciences, Buckinghamshire, UK). Band density was quantified by ImageJ software (National Institutes of Health, Bethesda, Maryland, USA).
Measurement of LPO by TBARS assay
Lipid peroxidation (LPO) was measured by thiobarbituric acid reactive substances (TBARS) assay. It is a well-recognized method for quantification of the lipid peroxides such as malondialdehyde (MDA). In this method, thiobarbituric acid (TBA) reacts with MDA (end products of LPO) to produce a pink color with maximum absorption at 532 nm. First, 500 μl of condition medium was added to 2.5 ml of Trichloroacetic acid (TCA) (20% (w/v)) and centrifuged at 1500g for 10 min to precipitate the proteins of the sample. Then, the precipitate was dispersed in 2.5 ml of sulfuric acid (0.05 M), and 2 ml TBA (0.2% in 2 M sodium sulfate) was added and incubated for 30 min in a boiling water bath. Finally, 4 ml n-butanol was added and absorbance was determined at 532 nm.
Measurement of total thiol molecules
To measure the plasma total sulfhydryl level, 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) was used as a reagent. This method is based on DTNB reaction with thiol molecules that forms a yellow complex, with a maximum absorbance at 412 nm. In brief, 0.2 ml of supernatant was added to 0.6 ml of tris(hydroxymethyl)aminomethane (Tris)–ethylenediaminetetraacetic acid (EDTA) buffer (Tris base 0.25 M, EDTA 20 mM, pH = 8.2), followed by the addition of 40 μl DTNB (10 mM) in methanol.Then, the test tube was centrifuged at 3000g for 10 min at room temperature and ultimately absorbance of the supernatant was measured at 412 nm.
Measurement of TAP
Total antioxidant power (TAP) was assessed by the ability of the cells to reduce ferric tripyridytriazine to a blue ferrous complex established as the ferric-reducing antioxidant power (FRAP) test. In this assay, the stock solution was prepared by mixing 300 mM acetate buffer (pH 3.6) with 16 ml acetic acid per liter of buffer solution, 10 mM 2, 4,6-tripyridyl-s-triazine (TPTZ) in 40 mM hydrochloric acid and 20 mM ferric chloride (FeCl3). Afterward, 10 μl of the MSC supernatant was added to 300 μl of the fresh working FRAP solution prepared by mixing 25 ml acetate buffer, 2.5 ml TPTZ solution, and 2.5 ml FeCl3 solution. Then the prepared reagent was warmed at 37°C and absorbance of blue color ferrous complex was measured by a microplate reader at 593 nm (BioTek ELX800).
Statistical analysis
The results are presented as mean ± standard error of the mean. Statistical significance was analyzed by one-way analysis of variance for multiple comparisons and unpaired Student’s t test for comparisons among two groups. Statistically, differences were assumed significant at p < 0.05.
Results
BM-MSC characterization
BM-MSCs were isolated from tibia and femur of the rats and cultured under specified conditions. The purified BM-MSCs displayed a uniform fibroblast-like appearance during the culturing process. At passage 3, CD markers of BM-MSCs were measured by flow cytometry, and the evaluations showed that the majority of the adherent cells (>95%) were positive for CD44, CD73, CD90, CD105, and CD106 but negative for CD31, CD34, and CD45 (a leukocyte marker) suggesting their mesenchymal origin (Figure 1(a) and (b)).

FACS analysis for BM-MSCs characterization. BM-MSCs were (a) negative for CD31, CD34, and CD45 and (b) positive for CD44, CD73, CD90, CD105, and CD106. FACS: fluorescence-activated cell sorting; BM-MSC: bone marrow-derived mesenchymal stem cell; CD: cluster of differentiation.
E2 promoted proliferation of BM-MSCs
MTT reduction assay was performed to elucidate the effect of E2 on BM-MSCs proliferation. The results showed that E2 (1000 and 100 nM) significantly increased proliferation of BM-MSCs in a dose-dependent manner. The effect of E2 was time dependent from 24 to 48 h, with a peak of 140 ± 7.89% of control at 48 h application. The cells were then exposed to serum-free medium–containing increasing concentrations of E2 (1–1000 nM) for 24–48 h. The data show that SD reduced cell viability and E2 protected BM-MSCs against SD in a concentration-dependent manner. The 24 and 48 h SD insult induced 30% and 40% cell death in BM-MSCs, respectively, but treatment with E2 (100 and 1000 nM) significantly enhanced survival of BM-MSCs (Figure 2).
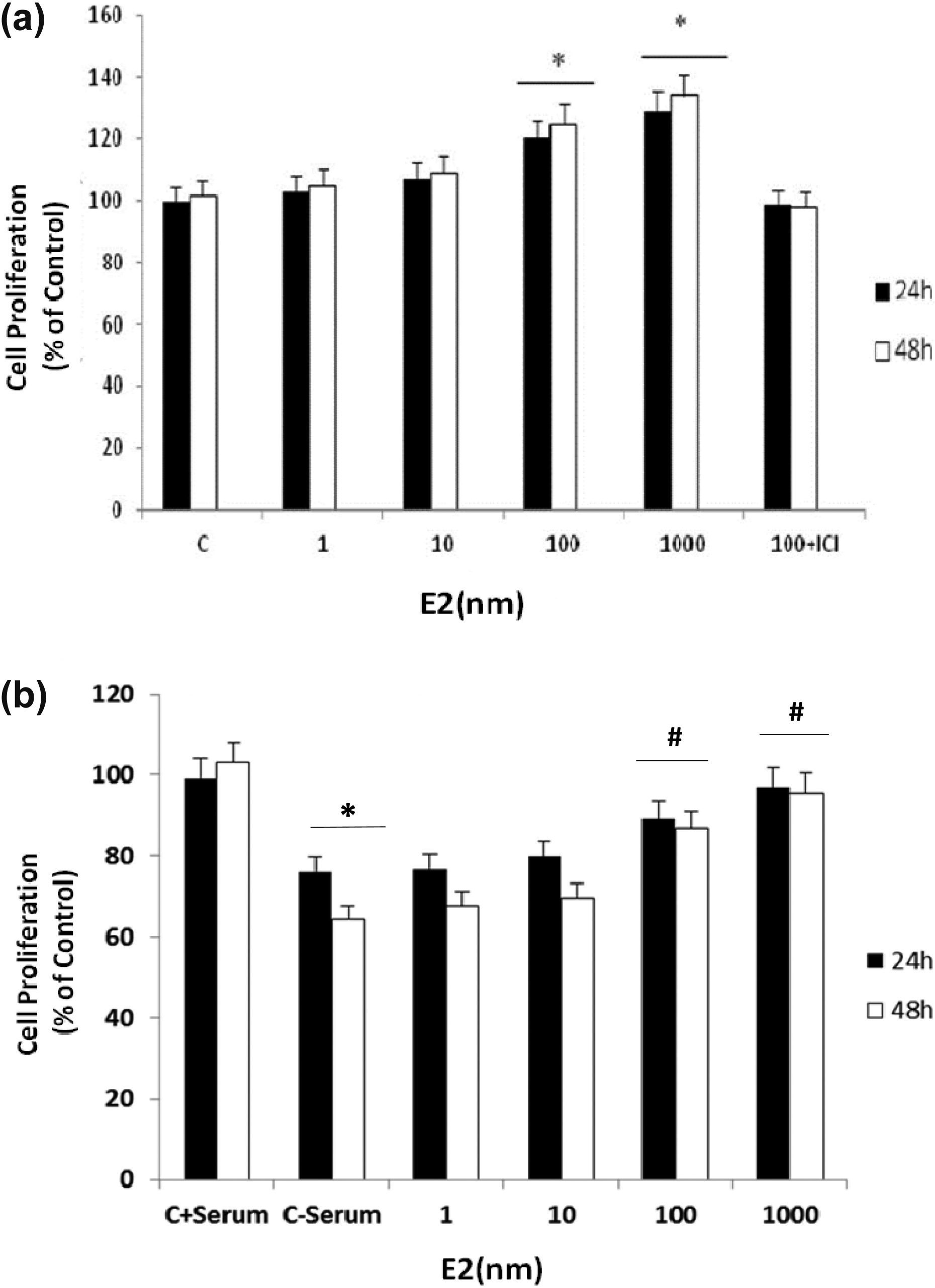
Effect of E2 on the viability of BM-MSCs. Cell viability was determined by MTT colorimetric assay. BM-MSCs were treated with different concentrations of E2 (1–1000 nM) for 24 and 48 h in the presence or absence of serum. (a) MTT results showed that the viability of BM-MSCs increased by E2 (100 and 1000 nM) after 24 and 48 h incubation and that of (b) E2 decrease the cytotoxic effects of SD on BM-MSCs (n = 8 in each concentration). Each experiment was repeated three times. C: control. Values are mean ± SEM. *p < 0.05: versus C + serum, #p < 0.0 5: versus SD group. E2: 17 b-estradiol; BM-MSC: bone marrow-derived mesenchymal stem cell; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SD: serum deprivation; SEM: standard error of mean.
E2 protected BM-MSCs from SD-induced apoptosis
PI staining assay was conducted to determine the effect of SD on cell survival. Cell viability was reduced by 24 h of exposure to SD compared with what was seen in cell culture that contained 15% serum. Treatment with E2 decreased apoptosis of BM-MSCs in a dose-dependent manner (Figure 3).

Inhibitory effect of E2 on BM-MSCs apoptosis. Flow cytometry after staining with PI was performed to determine BM-MSCs apoptosis. Each experiment was repeated three times.
E2 treatment modulates Bcl-2, Bax, and caspase 3 protein levels
Western blot analysis was performed to investigate the influence of E2 on Bax/Bcl-2 ratio and caspase 3 protein expression in BM-MSCs. Figure 4 shows that E2 significantly reduced SD-induced increased Bax/Bcl-2 ratio and decreased caspase 3 protein expression.

Effect of E2 treatment on antiapoptotic genes expression in BM-MSCs. BM-MSCs were exposed to serum-free medium with increasing concentrations of E2 (10 and 100 nM) for 24 h and then harvested. Total protein was extracted and expression of (a) Bax/Bcl-2 ratio and (b) caspase-3 was measured by Western blotting. β-actin was used as internal control. Values are mean ± SEM. **p < 0.01: versus C + serum, ***p < 0.001: versus C + serum,
E2 conditioning decreased ROS production
DCF-DA was used to detect intracellular accumulation of ROS. In the presence of ROS, dichlorodihydrofluorescein is oxidized to the highly fluorescent DCF by cellular esterase. In this assay, ROS production increased in BM-MSCs that were exposed to SD. From Figure 5, conditioning of the cells with E2 (10 and 100 nM) markedly decreased DCF fluorescence signal.

Effect of E2 on SD-induced ROS production in BM-MSCs. ROS was detected using DCF-DA. Each experiment was repeated three times. Values are mean ± SEM. ***p < 0.001: versus C + serum, ###p < 0.001: versus SD group. E2: 17 β-estradiol; BM-MSC: bone marrow-derived mesenchymal stem cell; SD: serum deprivation; SEM: standard error of mean; ROS: reactive oxygen species; DCF-DA: 2′-7′-dichlorodihydrofluorescein diacetate;
E2 conditioning modulates oxidative stress biomarkers
An increase in LPO and a decrease in total thiol molecules (TTMs) and TAP were seen in SD group. As shown in Figure 6(a), conditioning of BM-MSCs with E2 could significantly reduce LPO when compared with the SD group. Also a significant increase of TTM and TAP level was observed following treatment of cells with estradiol in serum-deprived groups (Figure 6(b) and (c)).

Changes in oxidative stress biomarkers in BM-MSCs exposed to SD and E2 for 24 h. (a) LPO, (b) TTM, and (c) TAP. Each experiment was repeated three times. Values are mean ± SEM. *p < 0.05. BM-MSC: bone marrow-derived mesenchymal stem cell; SD: serum deprivation; SEM: standard error of mean; TTM: total thiol molecules; TAP: total antioxidant power.
Discussion
This study was performed to determine whether E2 can protect BM-MSCs against SD-induced oxidative stress and apoptosis and also to elucidate major mediators involved. There are compelling evidences indicating that cell therapy has attracted interests as a future restorative treatment for many diseases. 24,25 BM-MSCs are the most frequently used cell type in regenerative therapies, and their unique characteristics make them a favorable candidate for cell transplantation purposes. 1 Despite all the promises, the major obstacle in cell therapy is the poor and insufficient viability as well as the low number of transplanted cells reaching the target tissues that greatly limit their potential therapeutic effects. It is well known that ROS is involved in induction of apoptosis, and based on this background, we hypothesized that agents with antioxidant activity have the capacity to scavenge ROS and as a result prevent cellular damages. In the present study, we found that E2 exerts cytoprotective effect against SD-induced injury by scavenging intracellular ROS and decreasing Bax/Bcl-2 ratio. Our results suggested that E2 may be utilized as a complementary strategy to improve the survival of BM-MSCs in transplantation studies.
Apoptosis is a multistep process that occurs by alteration in mitochondrial permeability, cytochrome c release, and subsequent activation of caspase cascade resulting in cell death. 26 Regulation of apoptosis is a complicated procedure that involves a number of pro- and antiapoptotic genes such as Bcl-2 and Bax. 27,28 It was shown that Bcl-2 is an antiapoptotic protein exists in the inner membrane of mitochondria, which protects cells from death and its overexpression, and suppresses initiation of apoptosis in response to various stimuli such as ischemia. 29 On the other hand, Bax is a proapoptotic protein that can form membrane pores, release proapoptotic factors such as cytochrome c, and activate the caspase cascades. 30
Serum withdrawal is a well-established model of induction of apoptosis that results in cell death by activating intrinsic/mitochondrial apoptotic pathway. 9,31 Moreover, it was shown that ROS generation is an important step in cell death induced by SD. 32 The current results showed that E2 conditioning could decrease SD-induced elevated ROS concentration in BM-MSCs. Our finding is in agreement with the previous studies indicating that E2 suppresses mitochondrial ROS production in endothelial cells and brain of male and female rats. 33,34 It was suggested by Stirone et al. that E2 exerts its vascular protection by modulating mitochondrial function and decreasing ROS production. 35
LPO is another important mechanism that involved in the production of ROS. LPO is a process by which different oxidants, such as free radicals, attack lipids, especially polyunsaturated fatty acids resulting in cell damage. 36 It is important to consider that antioxidative defenses that prevent LPO protect cells against free radical damage induced by oxidative stress conditions. 37 Also thiol-containing compounds with sulfhydryl groups are antioxidants that protect cells against free radical-induced damages. 38
The current results showed that SD significantly increased TBARS level as an important biomarker of LPO. Our results indicated that E2 treatment inhibited the peroxidation of lipids and elevated TTMs in serum-deprived cells. It was shown that estrogens are powerful antioxidants, especially by preventing LPO. 39 In line with our findings, Ayres et al. demonstrated the protective effect of E2 on LPO and DNA damage. 40
Evidences also suggested that ROS directly or indirectly elevates the gating potential of mitochondrial pores and decreases cell viability by damaging lipids, proteins, and nucleic acids. 41 It was also demonstrated that SD significantly increased Bax/Bcl-2 ratio in different cell types. 31,42 Cell damage is reduced by antioxidant defenses that neutralize produced ROS 43 and estrogens showed a 2.5 times more radical scavenging activity than vitamins E and C. 16,17 Current results showed that conditioning with E2 decreased the ratio of Bax/Bcl-2 by blocking SD-induced elevation of Bax and lowering Bcl-2. These results are in agreement with previous studies reporting that E2 has protected hepatocytes against hypoxia-induced apoptosis by upregulation of Bcl-2 expression and reducing ROS-dependent mitogen-activated protein kinases (MAPK) activity. 15 Moreover, it has also protective effect against homocysteine-induced apoptosis by upregulating Bcl-2 expression, attenuating cytochrome c release, and decreasing apoptotic cascade activation (Bax, caspases 3 and 9) in human dopaminergic SH-SY5Y cells. 44
The mechanism of SD-induced ROS generation is not fully understood. It was suggested by Lee et al. that mitochondrial ROS modulator 1 may be the key player involved in ROS production under SD. 45 It was also previously reported that estradiol can function as a potent antioxidant. 46 In oxidative stress situations, antioxidants can attenuate ROS accumulation and MAPK activation, indicating ROS involvement in the activation of MAPK signaling pathway. 47 Oxidative stress has been shown to stimulate MAPK pathway including c-Jun N-terminal kinases (JNKs)/stress-activated protein kinases (SAPKs) and p38 kinases. 48 Usually, the activation of JNK and p38 pathways is involved in apoptosis and growth arrest. 49 It was shown that E2 protected against hypoxia-induced cell injury in chicken hepatocytes through ER-mediated upregulation of Bcl-2 expression and also reducing the activity of ROS-dependent p38 MAPK, JNK/SAPK, and nuclear factor κB. 15 It was demonstrated that E2 suppressed JNK pathway and preserved transplanted human islet functional mass from pro-inflammatory cytokine-induced apoptosis. 50 It is also reported that E2 protects cardiomyocyte from apoptosis through inhibition of ROS production and regulation of p38 kinase pathway. 51
All these events lead to the activation of caspase 3, the final stage resulting in cell death and apoptosis. Despite the differing conditions under which cell injury can occur, caspase 3 activation is accepted universally as an apoptotic marker. 52 The reduction in caspase 3 protein level in the presence of E2 also supports previous results indicating that E2 protects the cells against apoptosis. 15,53 The results of this study demonstrate that E2 treatment provides a significant amount of protection against apoptosis signaling pathways that is induced by SD. In agreement with these results, it has been reported that E2 attenuates the ischemia-induced activation of caspase 3 and reduces DNA fragmentation. 54
In summary, our results revealed that serum withdrawal induces apoptotic cell death in BM-MSCs via mitochondrial pathway. In addition, E2 conditioning through different mediators attenuated oxidative stress, Bax/Bcl-2 ratio, and caspase 3 expression and therefore protected BM-MSCs against apoptotic insult. These results suggest that E2 may be applied as a promising therapeutic tool to promote cell survival and prevent apoptosis in stem cell-based therapies.
Footnotes
Conflict of interest
The authors declared no conflicts of interest.
Funding
This work was supported by a grant (number 22658) from Research Council of Iran University of Medical Sciences.