
Abstract
Zinc (Zn) has long been touted as a panacea for common cold. Recently, there has been some controversy over whether an intranasal (IN) zinc gluconate gel, purported to fight colds, causes anosmia, or loss of the sense of smell, in humans. Previous evidence has shown that IN zinc sulfate (ZnSO4) solutions can cause anosmia in humans as well as significant damage to the olfactory epithelium in rodents. Using an in vitro olfactory neuron model (the rat Odora cell line), we tested the hypothesis that Zn toxicity was caused by inhibition of the hydrogen voltage-gated channel 1(HVCN1), leading to acidosis and apoptotic cell death. Following studies to characterize the toxicity of zinc gluconate and ZnSO4, Odora cells were grown on coverslips and loaded with 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein acetoxymethyl ester to measure intracellular pH in the presence and absence of Zn salts. While we found that HVCN1 is not functional in Odora cells, we found that olfactory neurons in vitro maintain their intracellular pH through a sodium/proton exchanger, specifically the sodium proton antiporter 1. ZnSO4, at nontoxic levels, had no impact on intracellular pH after acute exposure or after 24 h of incubation with the cells. In conclusion, Zn toxicity is not mediated through an acidification of intracellular pH in olfactory neurons in vitro.
Introduction
In the 1930s, physicians, in an attempt to protect children from polio, treated several cohorts of children with intranasal (IN) ZnSO4 to ablate their olfactory mucosa. Clinically, the belief at the time was that the polio virus entered the body by inhalation and moved from the nasal cavity into the brain using the olfactory nerve as a conduit. Unfortunately, not only did the treatment fail to protect the children from polio but many of them also had a reduction or loss of their sense of smell. 1 Unfortunately, the adverse effect of ZnSO4 on the human olfactory system was “forgotten,” leading to another large problem, decades later.
With emerging evidence that oral Zn supplementation could shorten the duration of symptoms of the common cold, 2 a large number of Zn-containing products making claims to this effect were marketed. One of them, a zinc gluconate nasal gel, caused great concern among olfactory biologists and toxicologists. Predictably, individuals using the zinc gluconate nasal gel complained to their doctors and the US Food and Drug Administration (FDA) of impaired sense of smell 3 –5 . The product in question, Zicam nasal gel, was an over-the-counter product that was not regulated by the FDA. The FDA investigated the validity of claims linking Zicam nasal gel to impaired olfaction, and evidence supporting a link grew on the clinical side, as attempts to deflect concerns were mounted by the manufacturer and researchers funded by the company. 6 In 2010, a meta-analysis concluded that there was sufficient evidence that patient claims of hyposmia or anosmia resulting from the use of IN zinc gluconate formulation. 7 Altogether, combining the children receiving IN ZnSO4 treatments and the individuals using the Zicam nasal gel, intranasally administered Zn has damaged the sense of smell in thousands of humans.
The sensitivity of cells to Zn toxicity varies among cells and cell lines, and it is not known where olfactory neurons might fit into this spectrum. For example, at the low end of the toxicity spectrum, human pancreatic cell lines show “significant toxicity” at 10 μM, whereas the median lethal concentration (LC50) for Zn in the Ana-1 mouse macrophage cell line is approximately 150 μM. 8,9 For perspective, the free Zn concentration in the zinc gluconate nasal gel is approximately 35 mM, as described by Turner in his 2002 work. 10 There is also speculation in the literature that Zn salts may differ in their toxicity, with evidence presented that zinc gluconate is less toxic to nasal epithelia than ZnSO4. 6
Zn is an essential metal, yet a surprising gap exists in our knowledge of what happens to cells when they are exposed to high concentrations of Zn. In fact, data on how Zn might cause cell death are sparse and sometimes contradictory, as summarized in Table 1. Because of the observation that the proton channel hydrogen voltage-gated channel 1(HVCN1; also known as voltage-sensing domain only protein, VSOP), is highly expressed in the respiratory tract and is inhibited by zinc, 12,23 we tested the hypothesis that extracellular Zn inhibits HVCN1, causing intracellular acidosis and neuronal apoptosis. HVCN1 function is critical in the normal acid secretion process of respiratory tract epithelia and also in normal neutrophil function. 23,24 Therefore, the two goals of this study were to (i) determine whether zinc gluconate and ZnSO4 differ in their toxicity to the olfactory neuronal cell line Odora and (ii) to determine whether Zn exposure is associated with decreased intracellular pH in Odora cells.
Proposed mechanisms for zinc toxicity.

HVCN1: hydrogen voltage-gated channel 1; ATP: adenosine triphosphate.
Methods
Cell viability
Odora cells were obtained from Dr Dale Hunter and grown according to published protocols. 25 Odoras are an excellent model for the proposed studies because, as with the intact olfactory mucosa in vivo, the cells can be grown as immature neurons or as mature neurons (Table 2). To answer the first question concerning the contribution of the sulfate and gluconate anions to the olfactory toxicity of the respective Zn salts, immature Odora cells were seeded in 96-well plates (25,000 cells well−1) and allowed to attach and grow for 24 h. Cells were then treated with the respective Zn or sodium (Na) salts at concentrations ranging from 10 μM to 300 μM (based on preliminary range-finding studies). Mature Odoras were treated under identical conditions, except that the initial cell density was 10,000 cells well−1 since the cells grow more slowly than the immature cells and were given 48 h to spread out prior to treatment with the respective Zn and Na salts. After 24 h of treatment, crystal violet was used to assess viability as previously described 27 by measuring absorbance at 540 nm. Results were expressed as a percentage of control (water-treated) cells.
Growth conditions for Odora cells. 25

DMEM: Dulbecco’s modified eagle’s medium; FCS: fetal calf serum; OMP: olfactory marker protein; GAP-43: growth-associated protein.
HVCN1/VSOP expression and function
Total RNA was isolated from Odora cells, rat olfactory epithelium, and rat kidney using Tri Reagent (MRC, Cincinnati, Ohio, USA) according to the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized using the Verso cDNA kit (ThermoFisher, Pittsburgh, Pennsylvania, USA). Odora cDNA was amplified with primers specific for HVCN1 28 as well as other cell membrane transporters that are known to regulate intracellular pH, namely the Na+/H+ (proton) family of exchangers (NHE1–4; encoded by the SLC9A gene family). 29
Primer sequences
HVCN1: F 5′-TCGAAGCTCTAGGGCTGCTG-3′
R 5′-AGATCTGCCGTTCTGAGCGT-3′
NHE1: F 5′-TCTGCCGTCTCAACTGTCTCTA-3′
R 5′-CCCTTCAACTCCTCATTCACCA-3′
NHE2: F 5′-GCAGATGGTAATAGCAGCGA-3′
R 5′-CCTTGGTGGGGGCTTGGGTG-3′
NHE3: F 5′-GGAACAGAGGCGGAGGAGCAT-3′
R 5′-GAAGTTGTGTGCCAGATTCTC-3′
NHE4: F 5′-GGCTGGGATTGAAAGATGTATGT-3′
R 5′-GCTGGCTGAGGATTGCTGTAA-3′
Intracellular pH monitoring was conducted essentially as previously described by Amlal et al. 30 Briefly, cells (200,000 mL−1) were grown for 3 days on 3.1 × 1.3 cm2 glass coverslips in 60 × 15 mm2 polystyrene petri dishes. For “chronic” exposures, cells were treated with 0–150 µM ZnSO4 for 24 h. For “acute” exposures, cells were treated with ZnSO4 in the perfusion media. Cells were loaded with the fluorescent pH sensitive dye 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) for 10 min and then washed. Coverslips containing cells were positioned diagonally in a cuvette and placed in a thermostatically controlled holding chamber (37°C) in a Delta Scan dual excitation spectrofluorometer (Photon Technology Innovation, Edison , New Jersey, USA) and perfused with the appropriate solution (Table 3). The fluorescence ratio at excitation wavelengths of 500 and 450 nm (F 500/F 450) was used to monitor intracellular pH. Emission wavelength was 525 nm.
Solutions used in pHi studies 30 .
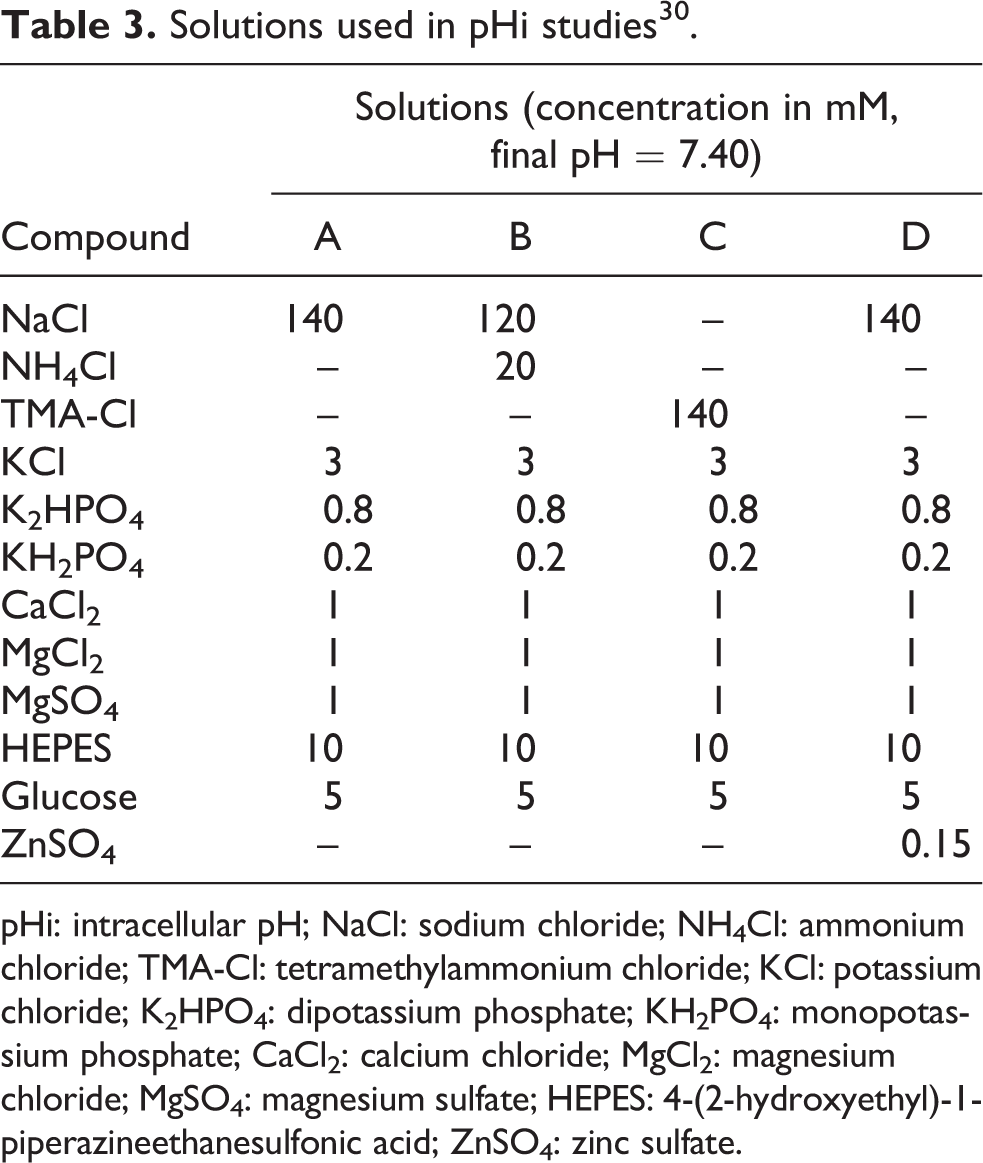
pHi: intracellular pH; NaCl: sodium chloride; NH4Cl: ammonium chloride; TMA-Cl: tetramethylammonium chloride; KCl: potassium chloride; K2HPO4: dipotassium phosphate; KH2PO4: monopotassium phosphate; CaCl2: calcium chloride; MgCl2: magnesium chloride; MgSO4: magnesium sulfate; HEPES: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; ZnSO4: zinc sulfate.
Results
Cytotoxicity of Zn and Na salts in Odora cells
As shown in Table 4, there is minimal difference between the 24 h LC50 values for zinc gluconate and ZnSO4. Importantly, as shown by comparison with the corresponding Na salts, toxicity is associated with the Zn ions, not the respective anions.
LC50 determinations for 24 h in mature and immature Odora cells.

LC50: median lethal concentration.
a100% viability at 300 μM.
bApproximately 75% viability at 300 μM.
Proton transporter expression in Odora cells
Reverse transcription polymerase chain reaction (RT-PCR) confirmed the expression of HVCN1 in rat olfactory mucosa and Odora cells (Figure 1). Of the NHE genes, only NHE1 was expressed (Figure 1; negative data not shown).

Top: PCR results of amplification with rat HVCN1 primers on the following cDNAs: lane 2; rat OE, lanes 3 and 4; rat nasal respiratory epithelium, lane 5; rat kidney (positive control), lanes 6 and 7; Odora, lane 8; rat OE, and lane 9; no cDNA (negative control). Bottom: PCR results of amplification with rat NHE1 primers on the following cDNAs: lane 2; no cDNA (negative control), lane 3; Odora, lane 4; Odora (no reverse transcriptase), and lane 5; rat kidney. PCR: polymerase chain reaction; HVCN1: hydrogen voltage-gated channel 1; cDNA: complementary DNA; OE: olfactory epithelium; NHE1: sodium–proton exchanger 1.
Proton transport in Odora cells
By RT-PCR, Odora cells clearly expressed the proton channel HVCN1. However, Odoras did not display Na-independent proton exchange activity, indicating that HVCN1 is not functional in Odora cells (Figure 2). In addition, proton transport via the NHE1 was not affected by treatment of Odora cells with Zn (Figure 3) and intracellular pH remained unchanged in Odoras treated either overnight or acutely with Zn salts (Figure 4).

Measurement of pHi in Odora cells. pH is indicated by the fluorescence ratio on the vertical axis. Cells are first equilibrated in a physiological buffer system (step 1, at left). Cells are then switched to buffer system with NH4Cl (step 2); the NH4Cl releases ammonia gas, which can pass into the cell without a transporter. The ammonia is oxidized by protons to re-form ammonium ions causing an increase in pHi. In step 3, cells are switched to TMA-Cl solution, which has no extracellular ammonium, resulting in the re-formation of intracellular ammonia that leaves the cell and excess intracellular protons leading to a sharp pH drop. If a proton channel (e.g. HVCN1) was active, there would be a recovery in pH since the cell is loaded with protons. However, in Odora cells, there is no pH recovery indicating that HVCN1 is not functional. Upon introduction of Na+ into the solution (step 4), pH recovers, indicating that Na+/H+ transporter is active (e.g. NHE1). Separate studies (not shown) indicate that NHE1 activity is not affected by extracellular zinc. pHi: intracellular pH; NH4Cl: ammonium chloride; TMA-Cl: tetramethylammonium chloride; HVCN1: hydrogen voltage-gated channel1; Na: sodium; NHE1: sodium–proton exchanger 1.

Representative trace of Odoras (step 1) treated with either ZnSO4 or water. pHi is indicated on the y axis, and time (in seconds) is indicated on the x axis. Following loading of cells with ammonium ions (step 2) to increase pHi, cells were perfused with TMA-Cl solution, inducing a dramatic decrease in pH (step 3). Cells were then switched to a Na+-containing medium (step 4), which initiated a rapid recovery of cell pH. The lack of difference in the final slope of the line (shown at the far right of each trace) indicates no effect of zinc on NHE1 activity. This result clearly indicates that Odora cells are devoid of Na+-independent H+ transport mechanisms even after severe cell acidification and that NHE1 is likely the only H+ transport mechanism expressed in these cells. ZnSO4: zinc sulfate; pHi: intracellular pH; Na: sodium; NHE1: sodium–proton exchanger 1; H+: proton.

Representative tracing to show no difference in pHi (y axis) in either zinc-free condition (initial 125 s) compared with pHi of undifferentiated Odora cells exposed to 100 μM ZnSO4 (125–350 s). Chronic (24 h) treatment with ZnSO4 (100 μM) was similarly without effect on pHi (not shown). Thus, although not all potential moderators of pHi were tested, these results indicate that zinc does not affect pHi. ZnSO4: zinc sulfate; pHi: intracellular pH.
Discussion
The purpose of these studies was twofold. First, to address a controversy in the literature concerning the toxicity of zinc gluconate versus ZnSO4 in the olfactory system of mice, we assessed the cytotoxicity of these salts in the rat olfactory neuronal cell line, Odora. There is some small difference in the toxicity of ZnSO4 and zinc gluconate salt solutions (Table 4), suggesting that the anions, while not cytotoxic themselves, may play a role in the toxicity of the respective salts. Due to its ability to chelate zinc, 31 it is possible that the Zn ion is less bioavailable as zinc gluconate than as ZnSO4. This suggests that there may be some credence to previous claims that zinc gluconate is less toxic than ZnSO4; however, the concentrations of Zn causing significant toxicity (LC50 values) in Odora cells is miniscule compared with the level of zinc gluconate in the Zicam nasal gel formulation, which is reported to be 35 mM zinc gluconate. 10 While the Zicam nasal gel formulation no longer contains Zn, homeopathic medicine Web sites continue to offer zinc gluconate nasal gel formulations 32 , so the problem has not disappeared altogether.
In addition to resolving questions around the relative toxicity of Zn to olfactory neurons, our second goal was to evaluate a potential mechanism by which Zn might cause cytotoxicity to olfactory neurons. Based on literature showing that Zn can cause intracellular acidosis by inhibition of the proton channel HVCN1, we evaluated this endpoint as the possible mechanism of toxicity of Zn to olfactory neurons. 12 While PCR results suggested the expression of the HVCN1 proton channel in Odora cells (Figure 1), based on our functional assay, this proton channel is either not expressed at the cell surface or is nonfunctional in Odora cells. This question remains unanswered because of the lack of a high-quality antibody for use in Western blots or immunohistochemistry at the time of these studies. The absence of a functional HVCN1 proton channel and Na+-independent proton exchanger was inferred by the lack of recovery of intracellular pH when the Odora cells were acidified in the absence of Na+ ions. If the HVCN1 proton channel or Na+-independent proton exchanger was present, then the cells would have recovered to their initial intracellular pH—but this did not occur. PCR results indicated the expression of NHE1 in Odora cells (but not other NHEs examined), and the results suggest that the NHE1 exchanger is functional in both undifferentiated and differentiated Odora cells, since recovery to initial intracellular pH (pHi) was observed when the cells were perfused with a Na+-containing solution after acidification. Odora cells are also able to respond to alkalinization of intracellular pH via a HCO3 −/Cl−dependent transporter, whose identity is currently unknown. Due to the lack of effect on initial pHi recovery from 24 h incubation with ZnSO4 and acute exposure to 150 μM ZnSO4, the mechanism of Zn toxicity on Odora cells is not through an intracellular pH mechanism.
The responses of olfactory neurons to Zn are not well characterized, and many questions are currently unanswered. For example, it is known that other tissues adapt to superphysiological Zn concentrations by upregulating metallothioneins (MTs) to sequester zinc, 33 –35 as well as Zn transporters (ZnTs) to remove excess cytoplasmic Zn, either via Zn efflux or recompartmentalization of Zn. 36 –38 MT1 and MT2 have been reported to be present in mouse olfactory epithelium, but most immunoreactivity was found in supporting cells, not in olfactory neurons. 39 MT3 message has been localized in the neurons of the olfactory epithelium and olfactory bulb, but none of the MT isoforms was induced following exposure to mercury vapors in the olfactory epithelium. 40 Failure of Zn to upregulate MTs or ZnTs could result in increased levels of cytosolic Zn, leading to apoptosis. 21 A recent article by Polak et al. 41 demonstrated that this pathway is operative in Caenorhabditis elegans, as well, in that this organism upregulated phytochelatin synthase, which catalyzes the formation of a glutathione polymer that functions similarly to MT, in response to exposure to Zn nanoparticles in C. elegans. These responses, or lack thereof, will be the subject of future investigations in olfactory neurons exposed in vivo and in vitro in response to Zn exposure, given that we have ruled out intracellular acidification as the mechanism of toxicity in these studies.
Footnotes
Acknowledgments
The authors thank Dr Marina Galvez, Tracy Hopkins, Dr Mansi Krishan, and Dr Sarah Pixley for their valued assistance to carry out the studies.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.