
Abstract
The present study aims to explore the mechanism of quinazolinone analogue HMJ-38-induced DNA damage in endothelial cells in vitro. We attempt to evaluate the antiangiogenetic response utilizing human umbilical vein endothelial cells (HUVECs). Herein, the results demonstrated that HMJ-38 incubation triggered DNA damage behavior and showed a longer DNA migration in HUVECs based on the comet assay and the analysis of DNA agarose gel electrophoresis to contact DNA smears. We further gained to determine a marker of DNA double strand breaks, phosphorylated histone H2A.X (Ser139) (γH2A.X), in HMJ-38-treated HUVECs by flow cytometry and Western blotting assay. We consider that HMJ-38 has caused an increase in γH2A.X, and DNA damage seemed to mediate through DNA-dependent serine/threonine protein kinase (DNA-PK) binding to Ku70/Ku80 as well as advanced activated p-Akt (Ser473) and stimulated phosphorylated glycogen synthase kinase-3β (p-GSK-3β) conditions in HUVECs. Importantly, the effect of above DNA damage response was prevented by N-acetyl-
Introduction
DNA damage is produced by internal or external damaging agents and shows a major danger to the integrity of the cellular genome. 1,2 Cellular defense system against the threat of DNA damage response swiftly modulates many physiological processes by activation of DNA damage signaling. 3 DNA double strand break (DSB) is one of the occurrences of triggered DNA damage, which is induced by reactive oxygen species (ROS), ultraviolet (UV), and radiation chemicals, that is formed during the course of normal metabolism. 4,5 Importantly, given that histone H2A.X phosphorylation at Ser139 as a novel end point to detect DNA breaks yields over an extensive chromatin domain flanking the site of DSBs that forms foci recruiting proteins involved in DSB repair. 6,7
In addition, DNA damage behavior or replication stress might affect the cell’s signaling sensors and transducer effectors through suppressing cell cycle transitions and inducing apoptosis or reducing transcription, DNA repair, chromosomal rearrangements, and cancer. 8 DSBs are typically recognized by DNA-dependent serine/threonine protein kinase (DNA-PK) implemented to DNA damage action. DNA-PK holoenzyme is a serine–threonine protein kinase, and DNA end-bridging activity results in phosphorylation of several protein substrates. 9,10 Alternatively, DNA-PK binding subunit is known as Ku, a heterodimer of Ku70 and Ku80 formed during DNA damage and danger situation. 11 –14 It is well documented that DNA-PK catalytic subunit (DNA-PKcs) can be activated by not only free DNA ends but also phosphorylation of the subunits Ku or the autophosphorylation. 15 Moreover, DNA-PK is necessary for the phosphorylation of Ser473 on Akt (v-akt murine thymoma viral oncogene homologue/protein kinase B (PKB)), following to cause DNA damage stress. 15,16 Consequently, Akt seems to be one of the major substrates for DNA-PK in the context of DNA damage repair and subsequently prompts the phosphorylation of glycogen synthase kinase-3β (p-GSK-3β) at Ser9 site in GSK-3 enzymatic activity. 17,18
It is well known that angiogenesis is required for invasive tumor growth or metastasis to generate and develop new capillary blood vessels and vasculatures. 19 –21 The previous reports have shown that the drugs or compounds targeting the tubulin cytoskeletal dynamics have received considerably to suppress the endothelial cell cytoskeleton or disrupt microtubule architecture as well as facilitate the behavior of DNA damage and apoptotic death, eventually causing to destroy angiogenic vessel networks and tumor microvessels when tumor angiogenesis. 22 –24 We demonstrated previously that newly synthesized HMJ-38 (2-(3′-methoxyphenyl)-6-pyrrolidinyl-4-quinazolinone) can suppress tubulin polymerization and exerts cytotoxic properties in various tumor cell lines. 25,26 Importantly, HMJ-38 inhibits vascular endothelial growth factor-stimulated vascular formation in the Matrigel plugs of mice and possesses the reduction of microvessel sprouting from the rat aortic ring, demonstrating that HMJ-38 has an antiangiogenic achievement and effect. 27 However, no studies have investigated the antiangiogenetic actions of HMJ-38 against oxidative stress-triggered DNA damage in vitro and its detailed underlying molecular mechanism in endothelial cells. To investigate these topics or questions, we selected human umbilical vein endothelial cells (HUVECs), a pivotal and commonly used in vitro model for angiogenesis progression. Hence, the purpose of the current study not only postulates that HMJ-38 might affect HUVECs and could trigger DNA damage stress but also evaluates the levels of protein and cellular approaches in HUVECs in vitro.
Materials and methods
Chemicals and reagents
HMJ-38, with chemical structure as illustrated in Figure 1(a), was provided by Dr Mann-Jen Hour (School of Pharmacy, China Medical University, Taichung, Taiwan), who designed and synthesized 2-phenyl-4-quinazolinone derivatives as novel antimitotic agents as described previously. 26,28 Medium 200, Low Serum Growth Supplement (LSGS), and trypsin–ethylenediaminetetraacetic acid (EDTA) were obtained from Gibco by Life Technologies (Carlsbad, California, USA). Anti-p-H2A.X (Ser139), anti-histone H2A.X, and anti-p-GSK-3β (Ser9) were purchased from Cell Signaling Technology (Beverly, Massachusetts, USA). The immunoblot antibodies obtained are as follows: anti-DNA-PKcs and anti-Ku70/Ku80 were bought from GeneTex Inc. (Irvine, California, USA). Antibodies against Akt, p-Akt (Ser473), p-Akt (Thr308), and GSK-3β as well as all horseradish peroxidase-linked anti-rabbit or anti-mouse immunoglobulin G (IgG) secondary antibodies were purchased from Merck Millipore (Billerica, Massachusetts, USA). All chemicals and reagents were of commercial grade obtained from Sigma-Aldrich Corp. (St Louis, Missouri, USA) unless stated otherwise.

HMJ-38 reduces cell viability and exerts cytotoxicity in HUVECs. (a) Chemical structure of HMJ-38 (2-(3′-methoxyphenyl)-6-pyrrolidinyl-4-quinazolinone). (b) HMJ-38 decreased the percentage of viable HUVECs. Cells at a density of 1 × 104 cells/well were placed into 96-well plates and then incubated with HMJ-38 at a final concentrations of 0, 0.625, 1.25, 2.5, 5, 10, and 20 μM or vehicle (0.1% dimethyl sulfoxide) for 24 h. After that, cells from each treatment were determined utilizing MTT-based in vitro toxicology assay. All data are the representatives of mean ± SD of three independent experiments. *p < 0.05 is considered a significant difference followed by Dunnett’s test.
Cell culture
HUVECs were obtained from the ScienCell Research Laboratories (Carlsbad, California, USA) and routinely cultured in Medium 200 with LSGS and without antibiotics, antimycotics, hormones, growth factors or proteins in the incubator at 37°C under a humidified 5% (v/v) carbon dioxide and 95% (v/v) air for further experiments in this condition as mentioned previously. 27,29
MTT-based in vitro toxicology assay
HUVECs at a density of 1 × 104 cells/well seeded into 96-well plates were incubated with a serial dilution of HMJ-38 having the final concentrations of 0, 0.625, 1.25, 2.5, 5, 10, and 20 μM or 0.1% dimethyl sulfoxide served as a vehicle control for 24 h and then were assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay for reduction as mentioned elsewhere. 30,31 After the end of incubation, the supernatant was discarded, and subsequently a 100-μl MTT solution (500 μg/ml) was filled to each well for 4 h at 37°C. The addition of 100 μl of 10% (w/v) sodium dodecyl sulfate in 0.1 N hydrochloric acid (HCl) per well solubilized the formazan crystals, following to measure by absorbance at 570 nm with a microplate reader. Data are expressed as percentage of MTT reduction as compared to untreated control condition.
Assessment of DNA damage by the comet assay
HUVECs at a density of 5 × 104 cells/well seeded into 24-well plates were treated with 0, 0.5, 1, 5, and 10 μM of HMJ-38 for 24 h. At the end of incubation, cells were harvested for examining DNA damage using the comet assay (single cell gel electrophoresis) as described previously with some modifications. 32,33 In brief, glass slides were precoated with 70 μl of 0.5% (w/v) low-melting-point agarose and 0.5% (w/v) normal melting point agarose at room temperature to dry on a flat surface. About 1 × 104 cells/gel for each sample was centrifuged (500g, 5 min at 4°C), and the cell pellet was then suspended in 100 μl of warm (37°C) 0.5% low-melting-point agarose, following which 70 μl aliquots were placed onto a glass slide. Each slide was immediately overlaid with a cover slip for 3 min on ice and then removed it. Afterward, each slide was exposed to lysis buffer (2.5 M sodium chloride, 10 mM Tris-HCl, 100 mM sodium-EDTA, and 1% (v/v) Triton X-100, adjusted to pH 10 with sodium hydroxide (NaOH)) at 4°C for 1 h. The slides were then transferred to an electrophoresis tank having cold electrophoresis alkaline buffer (300 mM NaOH and 1 mM EDTA at pH 13) at 4°C for 20 min. The electrophoresis was performed at 30 V and 300 mA for 20 min before slides were removed and flooded with neutralization buffer (0.4 M Tris-HCl at pH 7.5) at 4°C for 15 min before being dried in methanol for 5 min. HUVECs in the slides were stained with propidium iodide (2.5 μg/ml) for 5 min, and the comet tails in three random fields from each slide were visualized and photographed using a fluorescence microscope. Comet tails tended to increase rapidly with the levels of damage that was acquired by CometScore Software (Tritek Corp., Sumerduck, Virginia, USA) and calculated from the head center. Comet length was quantified and expressed as fold of control as described elsewhere. 34
DNA agarose gel electrophoresis for examining DNA damage
HUVECs at a density of 5 × 104 cells/well seeded into 24-well plates were incubated with 0, 1, 5, and 10 μM of HMJ-38 for a 24-h challenge. After that, the cells were harvested and DNA was isolated to lyse in 500 μl lysis buffer (20 mM Tris (pH 8.0), 10 mM EDTA, and 0.2% (v/v) Triton X-100) at 4°C for 20 min. Afterwards, proteinase K was added at 50°C to digest overnight, followed by exposure to 50 μg/ml RNase-A at 37°C for 1 h. The solution of phenol–chloroform–isopropanol (24:25:1; v/v/v) was used to extract DNA that was then resuspended into 100 μl of Tris–Borate–EDTA buffer after being precipitated with 50% isopropanol with 20 μg/ml glycogen. Approximately, the volume of 20 μl DNA (1 μg/μl) was loaded in each well, and DNA gel electrophoresis was performed using 1.8% agarose. After ethidium bromide was used to stain the DNA, the gel was photographed under UV light as described previously . 34,35
TUNEL evaluation by fluorescence microscopy
HUVECs at a density of 1 × 105 cells/well seeded into four-well chamber slides were incubated with vehicle control alone and 5 μM HMJ-38. After a 24-h incubation, HUVECs were washed twice in phosphate-buffered saline (PBS; pH 7.4) and fixed in 3.7% (v/v) formaldehyde for 15 min at 4°C. Cells were then washed twice with PBS and existed in 0.1% Triton-X 100 for 15 min before being employed to detect apoptotic DNA breaks by terminal DNA transferase-mediated dUTP nick end labeling (TUNEL) assay, following the protocol provided by the manufacturer (In Situ Cell Death Detection Kit, Fluorescein, Roche Diagnostics GmbH, Roche Applied Science, Mannheim, Germany). Breaks in cellular DNA were subjected to determine using a fluorescence microscope within 2 h of staining and to measure the TUNEL-positive cells in three random fields from each slide as described elsewhere. 35,36
Immunofluorescence staining for γH2A.X foci by flow cytometry
HUVECs at a density of 5 × 104 cells/ml seeded into the 24-well plates were exposed to 0, 0.5, 1, and 5 μM of HMJ-38 for 24 h. Cells were then harvested and fixed in suspension with methanol-free formaldehyde in PBS for 15 min at 4°C. After centrifugation, the pellet was resuspended in 0.1% Triton-X 100 for 15 min before being blocked in 1% bovine serum albumin for 1 h. After that, anti-p-H2A.X (Ser139) mouse monoclonal antibody (1:250 dilution) was added at room temperature for 4 h incubation. Cells were then centrifuged and tagged with Alexa Fluor 488 goat anti-mouse IgG secondary antibody (Molecular Probe by Life Technologies) for 1 h at room temperature in the dark as described previously. 37,38 The fluorescence of Alexa Fluor 488 from individual cells induced by excitation with a 488-nm argon ion laser was measured by flow cytometry (BD FACSCalibur flow cytometer, BD Biosciences, San Jose, California, USA) and quantified by BD CellQuest Pro Software (BD Biosciences).
Protein levels by Western blotting analysis
Approximately HUVECs at a density of 2 × 105 cells/ml were treated with 0, 0.5, 1, and 5 μM of HMJ-38 for 24 h. The total protein was extracted using the PRO-PREP protein extraction solution (iNtRON Biotechnology, Seongnam-si, Gyeonggi-do, Korea), and protein concentration was determined by Bradford assay as described elsewhere. 34,39 Protein abundance of DNA double-strand breaks associated levels (p-H2A.X (Ser139), histone H2A.X, DNA-PKcs, Ku70/Ku80, p-Akt (Thr308), p-Akt (Ser473), Akt, p-GSK-3β (Ser9), and GSK-3β) were determined by immunoblot analyses as described previously. 27,36 The blot was reprobed with an anti-β-actin antibody as an internal control to equal protein loading. Quantification of the density of each band was carried out using NIH ImageJ software version 1.46.
Intracellular ROS production by flow cytometric analysis
HUVECs at a density of 5 × 104 cells/ml seeded into 24-well plates were treated with 0, 0.5, 1, 5, and 10 μM HMJ-38 for 2 h. Cells were harvested, washed twice, and resuspended in the volume of 500 μl of 10 μM 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, an indicator for hydrogen peroxide) at 37°C for 30 min. Flow cytometric analysis was applied to measure the 2′,7′-dichlorofluorescein (DCF) fluorescence for the level of ROS generation as prescribed previously. 36,40
Effects of specific inhibitors on DNA damage and cell viability altered by HMJ-38
HUVECs at a density of 5 × 104 cells/ml seeded into 24-well plates were pretreated with or without 5 mM N-acetyl-
Statistical analysis
Data are analyzed using one-way analysis of variance to examine the significance of differences in measured variables between control and treated group/groups followed by Dunnett’s test. Numerical values are shown by mean ± SD from three separate experiments for each assay, and a value of p < 0.05 was considered as statistically significant
Results
HMJ-38 reduces the viability and has cytotoxicity in HUVECs
HUVECs after treatment with HMJ-38 (0, 0.625, 1.25, 2.5, 5, 10, and 20 μM) for 24 h that were harvested to test the effect of cell viability on HMJ-38-induced cell death. The cytotoxic effect of HMJ-38 on HUVECs was determined using MTT-based in vitro toxicology assay. As shown in Figure 1(b), cell viability is declined to 52.70 ± 3.86% after exposure to 5 μM HMJ-38 for a 24-h treatment. These results indicated that HMJ-38 concentration-dependently reduced the viability of HUVECs. Thus, the concentration of 5 μM HMJ-38 was chosen to investigate the molecular events in our following tests.
HMJ-38 provokes DNA damage and strand breaks in HUVECs
This study has shown that HMJ-38 can decrease cell viability and induce cytotoxicity in HUVECs (Figure 1(b)). To question whether nuclear DNA damage is involved in HMJ-38-reduced viability, the detections of comet assay, agarose gel electrophoresis, and TUNEL staining were used to analyze for HMJ-38-challenged HUVECs in vitro. Figure 2(a) as assessed using the comet assay indicates that a longer comet tail moment (DNA migration) occurred in a concentration-dependent increase in HMJ-38-treated samples, and control HUVECs only showed typical representative nuclei. As illustrated in Figure 2(b), we also discovered that quantification of comet length (fold of control) from damaged DNA was dramatically enhanced, which was measured by CometScore Software, and this effect in HMJ-38-treated HUVECs revealed in a concentration–response action. We further evaluated the influence of HMJ-38 on DNA breaks of HUVECs. We observed DNA smeared bands in gel electrophoresis in HMJ-38-treated HUVECs (Figure 2(c)). TUNEL assay also demonstrated that HMJ-38 at 5 μM also caused increased 3′-OH DNA ends of single- and double-strand DNA breaks in HUVECs and showed green fluorescence intensity. Alternatively, results from bright field indicated that cell morphological changes in apoptosis were found in HMJ-38-treated HUVECs as can be seen in Figure 2(d). Our data seem to indicate that exposure of HUVECs to HMJ-38 is capable of destroying DNA and inducing DNA strand breaks or apoptosis in vitro.
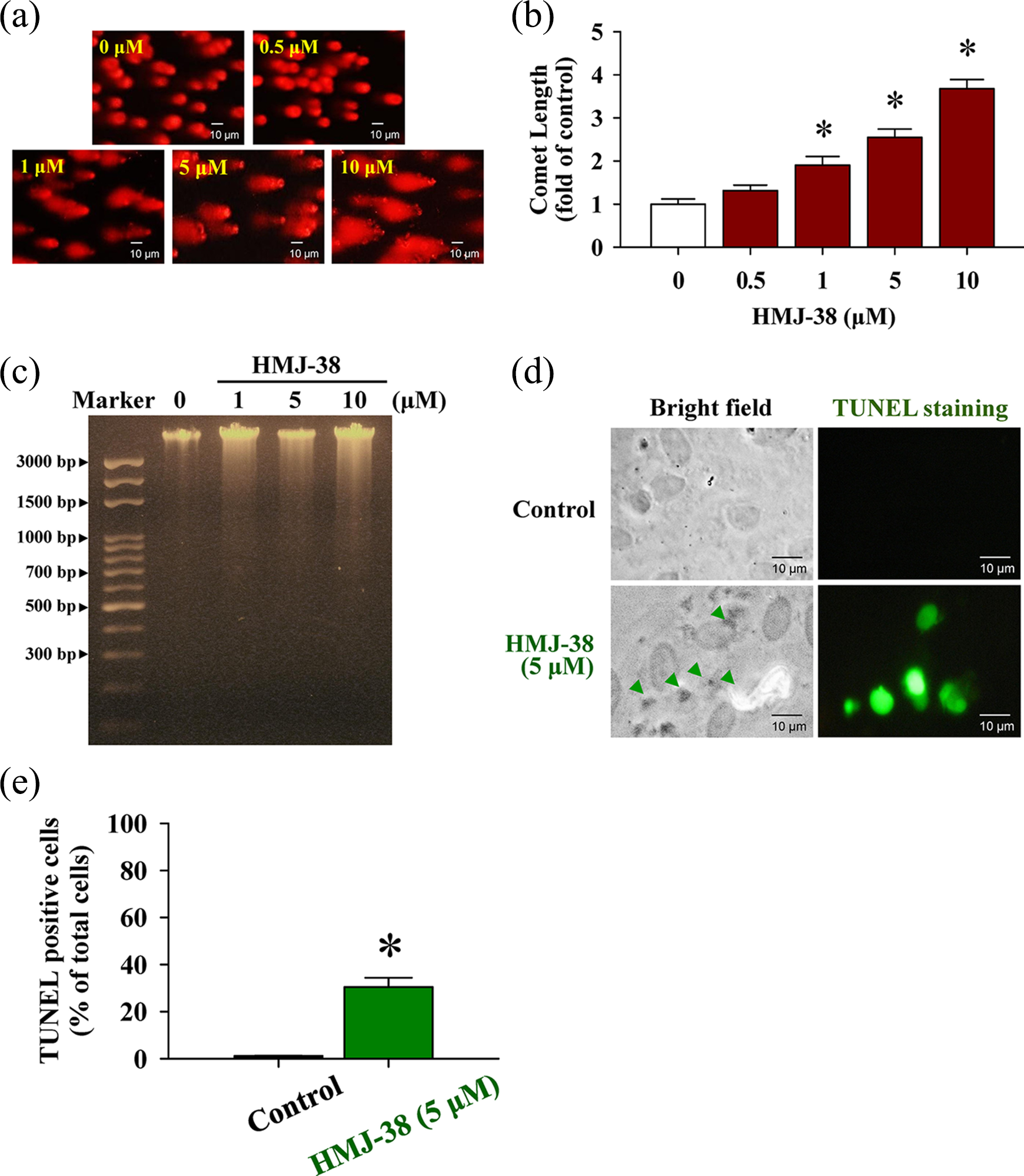
HMJ-38 treatment causes DNA damage in HUVECs. Cells were treated in the presence or absence of 0, 0.5, 1, 5, or 10 μM of HMJ-38 for 24 h. (a) DNA damage of HUVECs was triggered by HMJ-38, and a representative picture of comet tail from the comet assay were observed. (b) Quantitative values from comet length were measured by CometScore Software and each value (fold of control) is expressed. (c) The treated and untreated control cells were collected, and DNA was isolated from each treatment to determine the damaging DNA effect utilizing agarose gel electrophoresis. (d) TUNEL staining was applied to test DNA strand breaks. The symbol (▴) in the bright field indicated morphologic changes in apoptotic cells compared to the vehicle control. TUNEL-labeled cells (green color) revealed the experiencing DNA fragmentation or damage injury in HMJ-38-treated HUVECs. (e) TUNEL-positive cells were measured in three random fields from each slide. All data are repeated and representative of three independent experiments with similar results. Scale bar = 10 μm. The data were performed as mean ± SD of three separate experiments. *p < 0.05 is shown significant after analysis by Dunnett’s test. Each method was exploited as described in Materials and methods section.
HMJ-38 enhances γH2A.X foci generation and triggers DSBs in HUVECs
H2A.X represents histone H2A of the nucleosome core structure undergoing phosphorylation in response to DNA damage to form phosphorylated histone H2A.X (Ser139) (γH2A.X) foci which is considered a classic marker of DSBs formation. 6,41 HUVECs were incubated with 0, 0.5, 1 and 5 μM of HMJ-38 for 24 h to monitor the level of γH2A.X production. The flow cytometry detection/quantification assay revealed that the fluorescence was right shift to express HMJ-38-induced γH2A.X foci level in comparison to non-stimulated cells after we overlaid the profile using BD CellQuest Pro Software. The result seemed to show that the percentage of γH2A.X-positive cells peaked after HMJ-38 treatment (Figure 3(a)). Furthermore, the quantitative the fluorescent effect from flow cytometric analysis by BD CellQuest Pro Software was carried out on HMJ-38-challenged HUVECs, and we gained that HMJ-38 concentration-dependently promoted nuclei γH2A.X exposure in HUVECs (Figure 3(b)). In addition, this result was further substantiated by immunoblotting, and our findings showed that treatment of HUVECs with HMJ-38 resulted in the upregulation of the level of p-H2A.X (Ser139). The total level of histone H2A.X was slightly increased in HMJ-38-treated HUVECs (Figure 3(c)). These data demonstrated that HMJ-38 possessed the induction of DSBs in a concentration–response relationship.

HMJ-38 stimulates Ser139 histone H2A.X phosphorylation (γH2A.X) during DSBs in HUVECs. Cells at a density of 5 × 104 cells/well were exposed to 0, 0.5, 1, and 5 μM of HMJ-38 for 24 h. (a) H2A.X phosphorylation foci (γH2A.X) and function in response to DSBs that was determined by flow cytometry as described in Materials and methods section. Each cursor (↑) indicates the treated group. (b) In the bar chart, γH2A.X-labeled Alexa Fluor 488 was quantified utilizing BD CellQuest Pro Software. Results are plotted as mean ± SD of three independent samples. *p < 0.05 is expressed significant by Dunnett’s test. (c) Levels of p-H2A.X (Ser139) and histone H2A.X immunoblotting detection obtained from HUVECs treated or untreated with different concentrations of HMJ-38 as detailed in Materials and methods section. β-Actin was used to assess protein loading. Representative images are taken from three independent experiments with similar results.
Involvement of intracellular ROS generation stimulated by HMJ-38 in response to DNA damage in HUVECs
To elucidate whether ROS accumulation is required for HMJ-38-induced DNA damage response in HUVECs, cells after treatment with various concentrations (0, 0.5, 1, 5, and 10 μM) of HMJ-38 for 2 h were then determined for intracellular ROS level by flow cytometry using the H2DCFDA fluorescent probe. Our results shown in Figure 4(a) revealed the relative ROS production was a significant increase in HUVECs after exposure to HMJ-38 at 1, 5, and 10 μM. Moreover, pretreatment of HUVECs with 5 mM NAC (an ROS scavenger) markedly blocked DNA breaks and attenuated DNA migration (comet tail length) when compared with HMJ-38-treated alone sample (Figure 4(b)). We quantified the data using CometScore Software and showed that NAC pretreatment effectively eliminated comet length from HMJ-38-provoked DNA damage of HUVECs (Figure 4(c)). Owing to these findings, oxidative stress (ROS) plays an essential role in damaged DNA breaks and cell injury in HUVECs induced by HMJ-38.

Intracellular ROS generation contributes to HMJ-38-caused DNA damage in HUVECs. (a) Cells were incubated with 0.5, 1, 5, and 10 μM of HMJ-38 for 2 h. Assessment of DCF fluorescence index (ROS measurement) was piloted by flow cytometry, and quantitative values of DCF were expressed by utilizing BD CellQuest Pro Software. (b) After a 2-h pretreatment with or without 5 mM NAC, HUVECs were exposed to 5 μM HMJ-38 for 24 h. The damaged HUVECs were examined by the comet assay as described in Materials and methods section, and cells were protected by NAC after the comet tail (DNA damage) was cut back. The similar result was obtained at least from three repeat experiments. Scale bar = 10 μm. (c) CometScore Software was employed to quantify comet length (percentage of control) from damaged DNA of HUVECs. Each value is performed as mean ± SD of at least three samples. *p < 0.05 indicates a significant difference compared with the untreated control; #p < 0.05 value is significantly greater than that obtained for cells treated with HMJ-38 alone sample by Dunnett’s test.
Protein abundance of DNA-PK and Akt pathway is correlated with HMJ-38-triggered DNA damage in HUVECs
It is well documented that DNA-PKcs and Ku70/Ku80 form the core of a multiprotein complex (DNA-PK) that recruits to DSBs and promotes synapsis of the broken DNA ends. 13,42 To gain insights into the impact of DNA-PK and Akt molecule signaling in DNA breaks of HUVECs, we explored the possible mechanism and analyzed protein expressions in further detail. Cells were pretreated with or without NU7026 (a DNA-PK inhibitor) prior to incubation with 0, 0.5, 1, and 5 μM of HMJ-38 for 24 h. Results demonstrated that HMJ-38 significantly increased the protein level of DNA-PKcs and Ku70/Ku80 expression compared to untreated HUVECs (Figure 5(a)). We further focused on the downstream target of DNA-PK that phosphorylates Akt at Ser473 in response to nuclear DNA damage effect. 43,44 Alternatively, activated Akt phosphorylates GSK-3β at Ser9 residue to inactivate its kinase activity. 45 Our data displayed that there was an increase in phosphorylation of Akt at both Ser473 and Thr308 and p-GSK-3β (Ser9) protein expression in HMJ-38-treated HUVECs (Figure 5(b)). However, we also observed that the reduction of Akt in HUVECs after HMJ-38 exposure at 1 and 5 μM (Figure 5(b)). Therefore, we reasoned to further investigate the mechanism of DNA-PK function and followed that HUVECs were preincubated with NU7026 before HMJ-38 exposure. After that, we detected the DNA-PKcs and p-H2A.X (Ser139) protein expression in HUVECs utilizing immunoblotting analysis. As shown in Figure 5(c), HUVECs after pretreatment with NU7026 had less sensitive to DNA-PKcs and p-H2A.X (Ser139) signals than that of the HMJ-38-incubated cells. Moreover, HMJ-38-reduced viability in HUVECs was markedly abrogated when compared with HMJ-38 alone treated cells (Figure 5(d)). Correctively, these findings from our experimental approaches concluded that DNA-PK/Akt/GSK-3β pathway might contribute to ROS production-signalized DNA damage with DSBs in HUVECs in vitro.

DNA-PK and Akt/GSK-3β pathways are involved in HMJ-38-provoked DNA damage stress and cell death in HUVECs. Cells after exposure to 0, 0.5, 1, and 5 μM of HMJ-38 for 24 h were collected, and the total proteins were then prepared to detect immunoblots as described in Materials and methods section. Blot analyses for (a) DNA-PKcs and Ku70/Ku80 as well as (b) p-Akt (Thr308), p-Akt (Ser473), Akt, p-GSK-3β (Ser9), and GSK-3β were observed in cultured HUVECs. (c) To verify the influence of DNA-PK interaction, HUVECs were pretreated with or without 1 μM NU7026 (a DNA-PK inhibitor) for 2 h and further incubated with 5 μM HMJ-38 for 24 h exposure. NU7026 inhibited HMJ-38-upregulated DNA-PKcs and p-H2A.X (Ser139) protein expression by immunoblotting. All blots are carried out from three independent experiments with similar results. β-Actin served for loading control. (d) Cell viability was also determined by MTT method in the presence and absence of NU7026 after exposure to 5 μM HMJ-38 for 24 h in HUVECs. Data represent as overall mean ± SD of three independent experiments and were employed by Dunnett’s test. *p < 0.05 is recognized a significant difference compared with the value from the untreated control; #p < 0.05 is significantly different compared to HMJ-38-treated sample. p-GSK-3β: phosphorylated glycogen synthase kinase-3β.
Discussion
The current study is the first to successfully employ the comet assay, a high sensitive technique for DNA damage stress, 46,47 to clarify HMJ-38-induced DNA oxidative injury in endothelial cells. HUVECs are a good endothelial cell model and commonly used cell culture system for in vitro angiogenesis and anti-vascular action. The previously studies have proven that interfering with DNA metabolism and damaging DNA response by chemotherapeutic agents might be essential for treating cancer. 48,49 Additionally, evidence that DNA damage and apoptotic death of endothelial cells in tumor microenvironment have shown to destroy tumor microvascular networks during cancer angiogenesis and progression. 22 –24 It is worth notice that the antimitotic drugs or agents of which diverse binding sites on tubulin that can disrupt the angiogenic vessel or vascular endothelial cytoskeleton and inhibits tumor vasculature, leading to endothelial cell apoptosis and DNA damage in vitro. Also, these microtubule-binding drugs may inhibit cell proliferation by acting on the polymerization dynamics of spindle microtubules and hereafter cause endothelial cells injury to suppress angiogenesis. 50 –52
As depicted previously, HMJ-38 and 2-phenyl-4-quinazolinone derivatives have been synthesized to possess the microtubule-destabilizing effect and suppression of tubulin polymerization. 25,26 So far, regarding whether HMJ-38, the most potent agent for anticancer, 26 has DNA damage response and proapoptotic effects in endothelial cells is still unclear and not well investigated. Hence, this work programs to test the induction of HUVECs DNA damage and explores its signal transduction cascade in vitro. In this study, we observed that HMJ-38 treatment prompted DNA damage response and strand breaks in a concentration-dependent manner (Figure 2), resulting from the enhancement of HUVECs cell death and a loss of cell viability (Figure 1(b)). It is well-documented that γH2AX is a sensitive molecular marker of DSBs when there is a failure to repair DNA lesions, 6,7 which was detected by immunoblotting and immunostaining by flow cytometry in this study, which clearly indicated that DSBs in HUVECs are initiated by HMJ-38, which appeared after 24 h of incubation (Figure 3) and thereafter prompted the death process. These assays offered clear evidence of DSBs occurrence by HMJ-38 exposure. Thus, we decipher the contribution of HMJ-38-caused DNA damage behavior with DDBs, linking to the phosphorylation of histone H2A.X at Ser139 (γH2A.X) in HUVECs. The previous reports have also shown that the upregulation of ROS formation correlates with the sensitization of oxidative damage to DNA and modulates a network of executing molecular determinants. 2,53 To approach this question, the present study displayed that HMJ-38 greatly stimulated ROS production in HUVECs (Figure 4(a)). Importantly, NAC could quench ROS accumulation to diminish HMJ-38-mediated DNA damage in endothelial cells, which was measured by the comet assay (Figures 4(b) and (c)).
The proteins Ku70 and Ku80 are two critical regulatory subunits of DNA-PK that has been reported to play a vital role during the formation of DSBs to cause cell injury. 13,42,54 Furthermore, DNA-PK molecule is a signal of downstream effectors during DNA damage response by phosphorylating at carboxyl-terminus on the conserved Ser139 site flanking the breakage of histone H2A.X (γH2A.X). 55,56 Our pharmacological approach revealed that the protein expression of DNA-PKcs and Ku70/Ku80 was promoted in HMJ-38-treated HUVECs (Figure 5(a)). NU7026 consequently prevented the levels of DNA-PKcs and p-H2A.X (Ser139) from HMJ-38-induced HUVECs in vitro (Figure 5(c)). Data demonstrated that HUVECs after pretreatment with NU7026 efficiently increased cell viability in HMJ-38-challenged endothelial cells (Figure 5(d)). Notably, Akt/PKB is a family of three serine/threonine kinases and activated after the key residue at Ser473 phosphorylation (p-Akt (Ser473)), which can interact with DNA-PK signal. Additionally, GSK-3β function through phosphorylation at Ser9 is mediated to target Akt molecule, which enhanced cell apoptosis and DNA damage response. 15,17,18 In this study, we found that HMJ-38 induced an increase in p-Akt (Ser473) and p-GSK-3β (Ser9) levels but a decrease in Akt protein expression in HMJ-38-incubated HUVECs (Figure 5(b)). However, no significant alteration in GSK-3β level was observed following HMJ-38 treatment of HUVECs as shown in Figure 5(b). Intriguingly, p-Akt (Ser473) was also increased in treated cells (Figure 5(b)). Further work will fully verify the role of phosphorylated Akt in coordinating HMJ-38-modulated process in endothelial cells. This supports our hypothesis to show that HMJ-38 signalized DNA damage response with DSBs through DNA-PK/Akt/GSK-3β transduction cascase in HUVECs.
In conclusion, on the basis of our finding in the current study, HMJ-38 was proven to be involved in pharmacologic evidence regarding DNA damage effect and γH2A.X/DNA-PK signaling molecules in HUVECs as illustrated in Figure 6. To our knowledge, this novel insight is to elucidate that HMJ-38 might be developed for a newly synthesized therapeutic agent for antiangiogenetic effect during cancer treatment.

The schematic proposed diagram of oxidative stress-mediated DNA damage stress by HMJ-38 in HUVECs in vitro.
Footnotes
Authors’ Note
T.-H.L. and J.-G.C. contributed equally to this work.
Acknowledgements
The authors are grateful to Hsiu-Maan Kuo, PhD, and Ya-Ling Lin, PhD (Department of Parasitology, China Medical University) for providing partial constructs in this work.
Funding
This research was supported in part by the grant-in-aid award from the National Science Council (NSC), Republic of China (Taiwan) (J.-G.C.).