
Abstract
Aminophenol isomers (2-, 3-, and 4-aminophenols) are typically classified as industrial pollutants with genotoxic and mutagenic effects due to their easy penetration through the skin and membranes of human, animals, and plants. In the present study, a simple and efficient ultrasound-assisted emulsification microextraction procedure coupled with high-performance liquid chromatography with ultraviolet detector was developed for preconcentration and determination of these compounds in human fluid and environmental water samples. Effective parameters (such as type and volume of extraction solvent, pH and ionic strength of sample, and ultrasonication and centrifuging time) were investigated and optimized. Under optimum conditions (including sample volume: 5 mL; extraction solvent: chloroform, 80 µL; pH: 6.5; without salt addition; ultrasonication: 3.5 min; and centrifuging time: 3 min, 5000 rpm min−1), the enrichment factors and limits of detection were ranged from 42 to 51 and 0.028 to 0.112 µg mL−1, respectively. Once optimized, analytical performance of the method was studied in terms of linearity (0.085–157 µg mL−1, r 2 > 0.998), accuracy (recovery = 88.6– 101.7%), and precision (repeatability: intraday precision < 3.98%, and interday precision < 5.12%). Finally, applicability of the method was evaluated by the extraction and determination of these compounds in human urine, hair dye, and real water samples.
Keywords
Introduction
Aminophenol isomers including 2-aminophenol (2-AP), 3-aminophenol (3-AP), and 4-aminophenol (4-AP), have been widely used as raw materials or intermediates in the fields of pharmaceuticals, dyes, and so on. For example, these compounds are extensively used as couplers in oxidative hair dyes in combination with developers. Also, 4-AP is used in the production of paracetamol and could be formed by the degradation of paracetamol-containing medicines in the human body. 1,2 Nevertheless, APs are typically classified as industrial pollutants with genotoxic, mutagenic, and hepatotoxic effects due to their easy penetration through the skin and membranes of human, animals, and plants. 3,4 Therefore, determination of these compounds has become one of the most important subjects for biochemical and environmental analysis. 5 –9
Amount of APs in the environmental and biological samples is low and in order to determine these compounds by a suitable technique, an extraction or a preconcentration step is necessary. In the past few years, with the developing interest in a much simpler miniaturized configuration of extraction procedures, a novel liquid–liquid microextraction system named liquid-phase microextraction (LPME) was developed. 10 Because of rapidness, simplicity, and minimal use of solvent, LPME has attracted increasing attention and has successfully been applied to different types of sample. 11 –13 However, owing to some disadvantages such as relative long extraction times and instability of microdrop, relative low precision and sensitivity are often encountered.
In recent years, some modifications were made to overcome the above disadvantages and dispersive liquid–liquid microextraction (DLLME) was developed as a relatively new mode of LPME technique. 14 In DLLME, an appropriate mixture of extraction and dispersive solvents is dispersed as fine droplets in the sample solution and a cloudy solution is formed. DLLME is a simple, rapid, and an inexpensive procedure and provides high enrichment factors (EFs). However, the consumption of dispersive solvent can decrease the partition coefficient of analytes into the extraction solvent, which can limit the detection power.
More recently, it has been shown that ultrasound radiation can be used as a powerful means to produce very fine emulsions from two immiscible liquid phases. This leads to enlargement of interfacial contact surface between both the immiscible liquids, which can increase the mass transfer between the two phases. In this way, without the use of dispersive solvent, the ultrasound radiation can be used to disrupt the extractant phase in sample solution. This process is known as ultrasound-assisted emulsification microextraction (USAE-ME). The use of USAE-ME can lead to an increment in the sensitivity and extraction efficiency in a short period of time. 15,16 Also, the USAE-ME can be considered as a “green” extraction procedure because the consumption of organic solvent is reduced to the microliter level.
However, because of wide application of APs in industry as raw chemical materials and as important intermediates, the amounts of wastewaters from their production have been increasing. Moreover, as p-AP is a metabolite of aniline, nitrobenzene, and paracetamol, it can be used as a biological marker for human aniline exposure and other applications in occupational medicine and toxicology. 17 –19 It is also classified by the European Union as a mutagenic substance that may induce heritable genetic defects or increase their incidence. 20
As a result, a simple and rapid method for extraction and determination of APs in water, wastewater, and the human body is of great importance for both biomedical and environmental samples. The aim of the present study was to combine USAE-ME with high-performance liquid chromatography (HPLC) with ultraviolet (UV) detectors (HPLC-UV) and develop a simple and efficient method for determination of APs in human fluid and environmental water samples. To the best of our knowledge, this is the first report about the application of this procedure for the simultaneous determination of APs in the aforementioned samples. Therefore, effective parameters on the extraction efficiency were optimized and analytical performance of the method was evaluated. Finally, the developed method was successfully applied to analysis of human urine, hair dye, and real water samples and compared with normal DLME.
Experimental
Reagents and solutions
Aminophenol isomers (2-, 3-, 4-AP), HPLC grade acetonitrile and methanol, chlorobenzene (C6H5Cl), chloroform (CHCl3), dichloromethane (CH2Cl2), tetrachloride ethylene (C2Cl4), carbon tetrachloride (CCl4), sodium chloride (NaCl), and ultrapure water were all obtained from Merck Chemicals (Darmstadt, Germany). Sodium hydroxide and concentrated hydrochloric acid, bought from Merck Chemicals (Darmstadt, Germany) were used to adjust the pH of the samples. Commercial permanent hair dyes were collected from retail stores locally.
A mixture of stock solution containing APs at 1000 µg mL−1 was prepared in HPLC grade methanol. A series of standard solutions were prepared by mixing an appropriate amount of the stock solution with ultrapure water in a 10-mL volumetric flask. The aqueous solutions were prepared daily by diluting the standard mixture with ultrapure water. All the standard solutions were stored at 4°C in the dark.
Sample preparation
Urine
Two kinds of human urine samples were prepared from a healthy volunteer in our lab: (i) blank urine samples (APs-free) and (ii) model urine samples. Blank urine samples (APs-free) were prepared from the volunteer, who has not taken any drug at least for 1 week, whereas model urine samples were prepared at 3 and 6 h after the administration of paracetamol (750 mg single dose) and were stored for 1 week below 0°C in plastic containers. Before use, the samples were thawed at room temperature, and working solutions were prepared by diluting 4 mL of the urine sample in a 100-mL volumetric flask. 5 mL of the resulting solutions were filtered and subjected to the USAE-ME process.
Hair dye
Hair dye sample of 200 mg (±0.1 mg) was dissolved in ultrapure water in a 200-mL volumetric flask. Working solutions of hair dye samples were freshly prepared by diluting 2 mL of the stock sample in a 10-mL volumetric flask. 5 mL of the resulting solutions were filtered and subjected to the USAE-ME process.
Real water
Real water samples including tap, river, and waste water samples were collected from different locations of Iran. Then, 5 mL of water sample was subjected to the USAE-ME process, without any pretreatment.
All the standard solutions, solvents, and samples were filtered through a 0.45-µm membrane to eliminate particulate matters before analysis.
Apparatus
A Knauer HPLC system (Berlin, Germany), equipped with a K-1001 HPLC pump, D-14163 degasser, and a K-2600 UV detector, was used. Chromgate software (version 3.1) for HPLC system was employed to acquire and process chromatographic data. The analytical column contained an output delivery system III (ODS III; 250 × inner diameter 4.6 mm2, 5 µm; MZ-Analysentechnik, Mainz, Germany). The pH of the solutions was measured by a PHS-3BW model pH-meter (Bell, Italy). Fine droplets of organic solvent were obtained using a 50/60 kHz ultrasonic water bath (SW3, Switzerland), and an EBA20 model centrifuge (Hettich, Germany) was used to accelerate phase separation.
A mobile phase comprised of water/acetonitrile (70:30, v/v) at a flow rate of 0.80 mL min−1 was found to be optimum. Prior to use, the mobile phases were filtered through a 0.45-µm membrane filter and degassed under vacuum. The sample injection volume was 20 µL and the analytes were monitored at 235 nm (at room temperature).
USAE-ME procedure
Due to more complexity, one of the model urine samples (collected after 3 h and spiked with 50 ng mL−1 of AP standards) was selected for optimization process. Then, 5 mL of the pH-adjusted model sample without ionic strength adjustment was placed in a 10-mL glass centrifuge tube and 100 µL of extraction solvent (CHCl3) was injected into it. The tube was then immersed into an ultrasonic water bath, in such a way that the level of both liquids (bath and sample) was the same for 5 min of sonication. During the sonication, the solution became turbid due to the dispersion of fine extractant droplets into the aqueous bulk. The emulsion was centrifuged at 5000 rpm min−1 for 5 min and phase separation was occurred. The sedimented phase was removed from the bottom of the tube using a 100-µL HPLC syringe and transferred to another test tube. After evaporation, the residue was dissolved in 100 µL methanol and 50.0 µL was injected into the HPLC system for analysis.
Results and discussion
The effect of different parameters such as the type and volume of extraction solvent, pH and ionic strength of sample, and ultrasonication and centrifuging time were investigated. In this experiment, 5.0 mL of the model sample was used to study the extraction efficiency, based on peak area, under different experimental conditions. After optimization, the blank urine sample was used for the calculation of EF and recovery. The following equation was used to calculate the EF

where C sed and C 0 are the analyte concentration in the sediment obtained from the calibration graph of direct injection of standard solution and the initial concentration of analyte within the sample, respectively.
Optimization of USAE-ME procedure
Selection of extraction solvent
In the USAE-ME procedure, the appropriate selection of extraction solvent is of great importance, because of its effect on the emulsification phenomenon and extraction efficiency. An extraction solvent in this procedure has to meet several requirements: high affinity for compounds of interest, good emulsification efficiency in the aqueous sample, low solubility in water, and higher density than water.
Based on these criteria, 100 µL of CHCl3 (density 1.48 g mL−1), C2Cl4 (density 1.62 g mL−1), CCl4 (density 1.59 g mL−1), CH2Cl2 (density 1.33 g mL−1), and C6H5Cl (density 1.10 g mL−1) were studied. The results showed that CHCl3 had the highest extraction efficiency among the examined solvents. A lower surface tension of CHCl3 enables a higher cavitation under ultrasound radiation and hence a higher efficiency in the formation of emulsion is obtained. Therefore, CHCl3 was selected as an optimum extraction solvent for further optimization studies. Figure 1 shows the effect of the extraction solvent on the extraction efficiency.
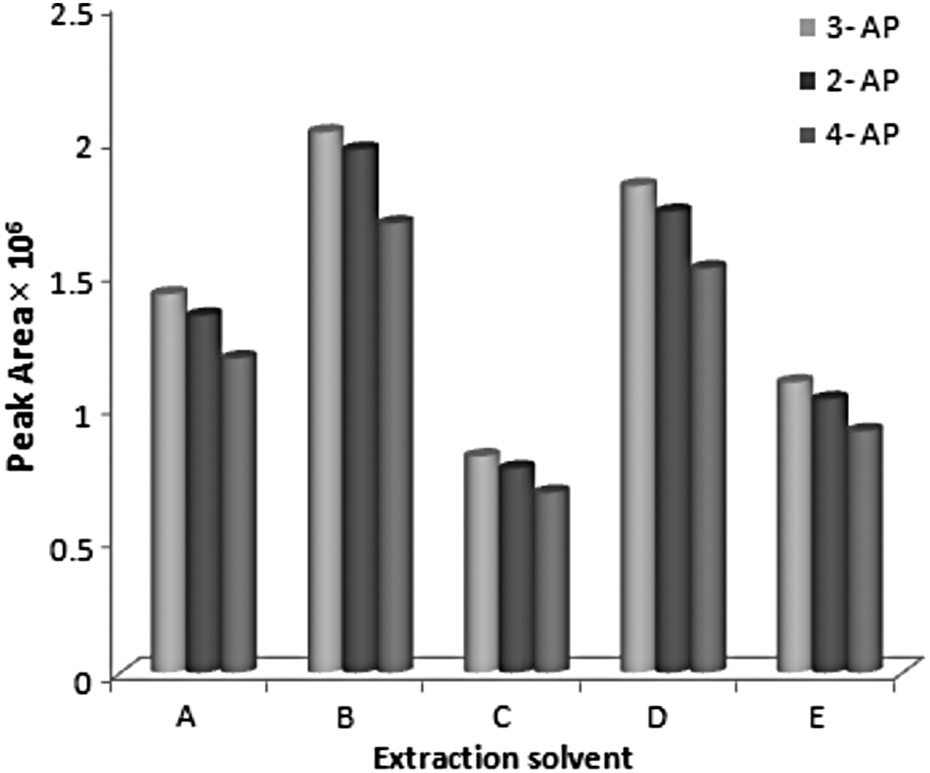
Effect of different extraction solvents on the extraction efficiency of APs. A: C2Cl4, B: CHCl3, C: CH2Cl2, D: CCl4, E: C6H5Cl. Extraction conditions: sample volume, 5.0 mL; extraction solvent volume, 100 µL; sonication time, 5 min; centrifugation time, 5 min; without salt addition.C2Cl4: tetrachloride ethylene, CHCl3: chloroform, CH2Cl2: dichloromethane, CCl4: carbon tetrachloride, C6H5Cl: chlorobenzene; APs: aminophenols.
Effect of the volume of extraction solvent
Since the volume of extraction solvent can influence the occurrence of the cloudy state of solution, it can play a key role in extraction efficiency. Besides, from the consideration of minimum consumption of solvent, the volume of extraction solvent should be as small as possible.
To study the effect of this parameter, different volumes of CHCl3 in the range of 40–120 µL were examined. The results are shown in Figure 2. As can be seen, when the volume of extraction solvent was increased, the extraction efficiencies were increased until 80 µL. At higher volumes from this volume, the extraction efficiency almost remained constant or slightly decreased. For the extraction volumes <40 µL, the sedimented phases were hardly removed by microsyringe and the reproducibility reduced drastically. Furthermore, some of the extraction solvents could not be well emulsified in the sample solution when their volumes were <40 µL. Therefore, 80 µL of CHCl3 was selected for further optimization experiments.

Effect of organic solvent volume on the extraction efficiency of APs. Extraction conditions: sample volume, 5.0 mL; extraction solvent volume, 100 µL; sonication time, 5 min; centrifugation time, 5 min; without salt addition. APs: aminophenols.
Effect of sample pH
Selection of sample pH is of great importance on the extraction procedure because the analytes can be better extracted in neutral molecule forms, which are favorable for the hydrophobic phase.
To investigate the effect of pH on the extraction efficiency of APs, the pH of the model sample was studied in the range of 2–7. The highest extraction efficiencies were obtained when the pH of sample was set at 6.5, at which the interested compounds had greater affinity for the extraction of solvent. Hence, pH 6.5 was used in the following experiments.
Effect of ionic strength
The presence of salt has different influences on the extraction efficiency of analytes in USAE-ME procedure. It is well known that the addition of salt can potentially decrease the solubility of the analytes in the aqueous phase and enhances their partitioning into the organic phase (salting-out effect), which improves the extraction efficiency. On the contrary, salt addition may change the physical properties of the Nernst diffusion film and reduce the diffusion rate of solutes into the organic microdrops (salting-in effect). Also, with increasing the viscosity and density of the medium due to salt addition, ultrasound radiations can be absorbed and dispersed as heating energy. This undesirable effect can prevent the organic phase from being dispersed into fine droplets and therefore the efficiency of emulsion formation can be drastically reduced. 21,22
In this experiment, the influence of ionic strength on the extraction efficiency of APs was investigated by adding different amounts of NaCl (0–30 mg mL−1) into the model sample. The salt addition has no significant effect on the extraction efficiency, perhaps because of the two opposite effects of salt addition in USAE-ME of APs. Hence, salt addition was not used in the subsequent experiments.
Effect of sonication time
It is clear that any parameter affecting dispersion phenomenon can alter the efficiency of extraction procedure. Dispersion is one of the most important stages in USAE-ME procedure. As the sonication time increases, the fraction of dispersed phase increases. This can lead to a greater surface contact between two phases and provide better extraction efficiency.
The effect of sonication time was evaluated in the range of 1–5 min. The results showed that sonication for at least 1 min was necessary to form a complete cloudy solution. As can be seen (Figure 3), the extraction efficiency was increased from 1.0 to 3.5 min and then only slightly changed when the ultrasonic time was more than 3.5 min. Therefore, this time was selected as the optimum ultrasonic time.
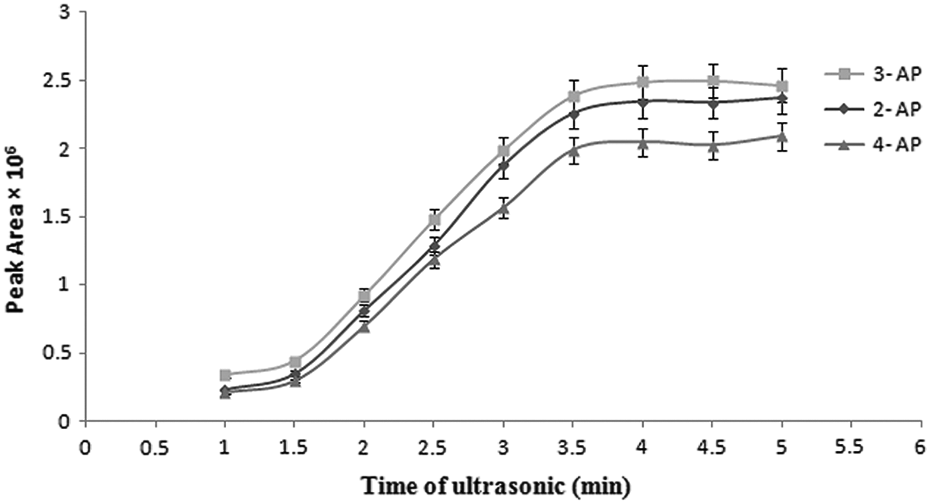
Effect of sonication time on the extraction efficiency of APs. Extraction conditions: sample volume, 5.0 mL; extraction solvent volume, 100 µL; sonication time, 5 min; centrifugation time, 5 min; without salt addition. APs: aminophenols.
Effect of centrifugation time
A good selection of centrifugation interval can insure satisfactory phase separation and sequentially lead to higher extraction efficiency. In general, a higher rate of centrifugation can lead to a shorter centrifugation time and better phase separation. So, the maximum rate of the centrifuge (5000 r min−1) was used in the experiments.
Centrifugation time in the range of 1–5 min was investigated and the best extraction efficiency was achieved at 3 min. The extraction efficiency was lower when the centrifugation time was too short, while longer times had no significant effect on the extraction efficiency. Therefore, 3 min was selected as centrifugation time.
Method performance
Analytical performance
Under the optimum conditions, the analytical parameters including limits of detection (LODs), linear dynamic ranges (LDRs), EFs, recoveries (R), and the repeatability (intraday and interday precision) were determined to evaluate the performance of the proposed method.
LODs were calculated as the minimum concentration providing chromatographic signals three times higher than background noise (Table 1). The linearity of the method was evaluated using mixed working solution with analytes over the concentration range of 0.085–157 µg mL−1. The calculated calibration curves gave a good linearity for all analytes with correlation coefficients higher than 0.9987 (Table 1).
Figures of merit for the USAE-ME of aminophenol isomers.a

EF: enrichment factor; APs: aminophenols; USAE-ME: ultrasound-assisted emulsification microextraction; r 2: squared correlation coefficient; LDR: linear dynamic range, LOD: limits of detection; RSD: relative standard deviation; ID: inner diameter.
aConditions: mobile phase water/acetonitrile (70:30, v/v); flow rate = 0.80 mL min−1; column ODS III (250 mm × ID 4.6 mm, 5 µm); injection volume = 50 µL; λ = 235 nm.
b n = 5.
Repeatability (intraday and interday precision) was evaluated by analyzing five replicates of model sample at three different concentration levels (low, middle, and high) on the same day and on five different days. As can be seen (Table 1), precision studies showed a suitable relative standard deviation for interested compounds. The EFs were calculated based on equation (1), and the results are shown in Table 1.
The accuracy of the method was confirmed by spiked recovery test. The recovery of the analytes, at three levels (0.0, 4.0, and 8.0 µg mL−1), were evaluated, and the results are shown in Table 2.
Mean recovery percentages and RSD for the determination of AP isomers at three concentration levels (n = 3).

APs: aminophenols; RSD: relative standard deviation; ID: inner diameter.
aConditions: mobile phase water/acetonitrile (70:30, v/v); flow rate = 0.80 mL min−1; column ODS III (250 mm × ID 4.6 mm, 5 µm); injection volume = 50 µL; λ = 235 nm.
bPeak area was used for quantification.
Analysis of real samples
To evaluate the applicability of the proposed method, three types of sample (human urine, hair dye, and water) were analyzed under optimum conditions. In order to eliminate possible matrix effects, the standard addition method was adopted for the quantitative determination of APs in the real samples. The concentrations of APs are shown in Table 3. The obtained results indicated that 4-AP can be one of the common pollutants in the environmental waters. It should be mentioned that the hair dyes were purchased from retail.
Determination of aminophenol isomers under optimum conditions (n = 3).a

APs: aminophenols; ND: not detected; ID: inner diameter.
aConditions: mobile phase water/acetonitrile (70:30, v/v); flow rate = 0.80 mL min−1; column ODS III (250 mm × ID 4.6 mm, 5 µm); injection volume = 50 µL; λ = 235 nm.
bPeak area was used for quantification.
The typical chromatograms for urine samples are shown in Figure 4. The presence of 4-AP in the urine sample 1 (peak 1 in chromatogram C) indicated that the 4-AP is the only isomer produced by the metabolism of paracetamol. With regard to the chromatograms, it can be seen that the sample matrices had no significant interferences for the determination of APs in the real samples.
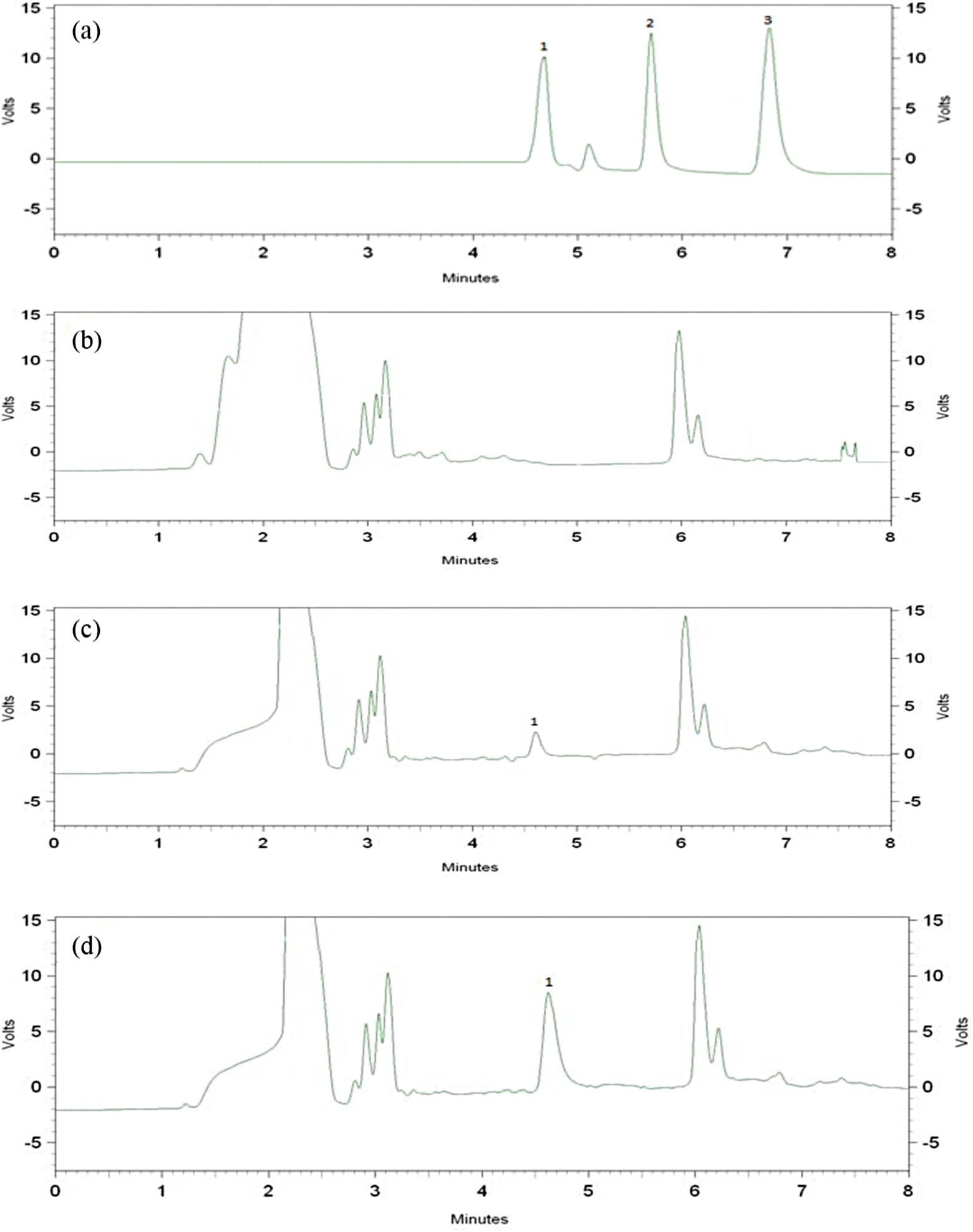
HPLC chromatograms of (a) standards (5 µg mL−1), (b) blank urine sample, (c) model urine sample, and (d) spiked model urine sample (3 µg mL−1 of 4-AP) after USAE-ME. Peak identification: (1) 4-AP, (2) 3-AP, (3) 2-AP. APs: aminophenols; HPLC: high-performance liquid chromatography; USAE-ME: ultrasound-assisted emulsification microextraction.
Comparison of the USAE-ME with normal DLLME
The applicability of normal DLLME (N-DLLME) procedure was also studied for the determination of APs in the real samples. Under optimum conditions (including sample volume: 5 mL; extraction solvent: CHCl3, 100 µL; dispersive solvent: tetrahydrofuran, 400 µL; pH: 6.5; without salt addition; and centrifuging time: 3 min, 5000 rpm min−1), the 4-AP was not detected in urine samples. The extraction efficiency and analytical performance of the N-DLLME was evaluated and the results showed the priority of USAE-ME procedure. Since satisfactory results were not obtained using N-DLLME procedure, they were not reported here.
The presence of 4-AP in the urine sample 1 showed that, if a suitable method such as USAE-ME could be applied to the APs preconcentration, it could improve the sensitivity of a common HPLC-UV for the determination of these compounds in the biological and environmental samples.
However, after APs preconcentration with USAE-ME method, if a more selective analysis technique such as liquid chromatography–mass spectrometry (LC-MS) could be used, it would be better for the identification and confirmation of the analytes in real samples. Meanwhile, LC-MS usually demands a tedious sample preparation and is complicated. Furthermore, LC-MS is not available everywhere and it is an expensive technique. According to the obtained results, the proposed method was sensitive enough for the determination of the APs in the model biological and environmental samples so that there was no need to use such expensive method. The proposed method was compared with other techniques and, as can be seen (Table 4), its analytical performance is comparable with, or better than, previous methods developed for determination of the APs in the similar samples.
Comparison of the proposed method with other methods, without advanced detection instruments such as mass spectrometry, which have been used for determination of the interested compounds.

LDR: linear dynamic range, LOD: limits of detection, EF: enrichment factor; R: recovery; HPLC: high-performance liquid chromatography; UV: ultraviolet; FID: flame ionization detector; LLE: liquid–liquid extraction; GC: gas chromatography; MEKC: micellar electrokinetic chromatography; USAE-ME: USAE-ME: ultrasound-assisted emulsification microextraction.
Conclusion
In the present study, an efficient USAE-ME procedure was developed for the extraction of AP isomers in different types of sample (human urine, hair dye, and real water samples) prior to HPLC-UV. The results indicated that USAE-ME can be used as a simple, precise and rapid method that has good recovery and EF for APs analysis. Also, the use of ultrasound irradiation to disrupt the extractant phase reduces the consumption of organic solvent, because the necessity of using a dispersive solvent is not needed. Furthermore, the common HPLC-UV was used for the simultaneous determination of target analytes in the complex real samples. The APs were collected from the end of HPLC column and confirmed by the ultraviolet absorption spectra separately. The results indicated that the APs were well separated and sample matrices had no significant interferences for the determination of APs in the real samples.
Footnotes
Funding
The authors would like to thank Semnan University Research Council for financially supporting this work.