
Abstract
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a neurotoxin associated with drug abuse and causes permanent symptoms of Parkinson's disease (PD) by destroying dopaminergic neurons in the substantia nigra of the brain. In the present study, the neuroprotective effects of two carboxylic acid compounds, viz. alpha-ketoglutarate (A-KG), a Kreb’s cycle intermediate and ethyl pyruvate (EP), a lipid-soluble analogue of pyruvate, were evaluated against MPTP intoxication in mice and compared with madopar (MD; combination of levodopa plus benserazide), a standard drug. Animals received oral treatment of A-KG (500 mg/kg), EP (100 mg/kg) or MD (5 mg/kg) daily for 5 days followed by intraperitoneal administration of MPTP (20 mg/kg) and posttreatment (+10 min) of A-KG, EP or MD daily for the remaining 5 days. MPTP caused the inhibition of complex I of electron transport chain accompanied by oxidative stress in the brain. It also caused cytotoxicity in the midbrain region as characterized by histology and immunohistochemistry. Treatments of A-KG and EP were found to resolve the loss of motor coordination, oxidative stress, diminished complex I activity and tyrosine hydroxylase–positive neurons in midbrain. A-KG and EP also regressed the histological damage in the brain and minimized the accumulation of alpha-synuclein in the midbrain region. The data suggest that A-KG and EP which are nontoxic carboxylic acid compounds could be of potential therapeutic value in the treatment of PD.
Introduction
Parkinson’s disease (PD) is a profound movement disorder resulting from progressive degeneration of the nigrostriatal dopaminergic pathway. Although its etiology remains unknown, emerging evidence suggests that the cause of PD is multifactorial, involving genetic predisposition, innate characteristics of the nigrostriatal dopaminergic system, and exposure to environmental toxins. 1,2 The finding that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) poisonings led to Parkinsonism 3,4 in drug addicts shifted the focus toward the environmental factors as potential PD initiators or contributors. Since then, MPTP model has been widely used to study the pathophysiology of PD and to develop novel antiparkinsonian therapies. MPTP is highly lipophilic, crosses the blood–brain barrier within minutes. 5 Once in the brain, the protoxin MPTP is oxidized to MPDP+ (1-methyl-4-phenyl-2,3-dihydropyridinium) by monoamine oxidase B in glia and serotonergic neurons, which is further oxidized to MPP+ (1-methyl-4-pyridinium). 6 The MPP+ thus generated sets off various cascade of events in neuronal cells, MPP+ increases oxidative stress, 7 initiates reactive oxygen species (ROS)-dependent cascades 8 and stimulates apoptotic cell death. 9 In the MPP+mediated signaling pathway, MPP+ not only regulates the activation of Akt, glycogen synthase kinase 3b, and extracellular signal-regulated kinase (ERK) 10 but also activates various factors related to cell death such as Bcl-2. 11
The first-line treatment for patients with PD is dopamine-replacement drug therapy. Perhaps the best known example of this therapy is levodopa, which remains the “gold standard.” Levodopa is decarboxylated to dopamine, both in the peripheral tissues and in the central nervous system (CNS). Hence, levodopa is given in combination with benserazide which acts as an inhibitor of peripheral decarboxyalses and allows dopamine to build solely in the brain. However, the use of levodopa as well as many other drugs has several limitations like their long-term use leads to major motor side-effects particularly dyskinesia and decreased efficacy. 12 Furthermore, most importantly, such treatments are largely symptomatic and do not prevent the pathology. 12,13 For this reason, there is a need to develop new treatments aimed at stopping or slowing down the progression of the disease. Indeed, there have been many studies over the years mainly in animal models of the disease, reporting some substances, or treatments (e.g. deep brain stimulation), 13 save or offer neuroprotection to the dopaminergic system in parkinsonian cases. One such substance is the indoleamine, melatonin.
Recently, pyruvate, a simple keto acid, has become popular for its antioxidant functions. Pyruvate detoxifies peroxynitrite (ONOO+), hydrogen peroxide (H2O2) and hydroxyl radicals in direct, nonenzymatic reactions and increases the formation of nicotinamide adenine dinucleotide phosphate via metabolism, thereby reducing glutathione disulfide to glutathione (GSH). 14 However, the usefulness of pyruvate as a therapeutic agent is limited by its very poor stability in solution. Ethyl pyruvate (EP) was developed as a more stable analogue of pyruvate for use as a component of an intravenous fluid administration. The antioxidant and anti-inflammatory effects of EP are similar to those of pyruvate. 15 –17 As an anti-inflammatory agent, EP inhibits nitrite/nitrate (NO) release as a proinflammatory marker and cytokines in many ischemia/reperfusion injury and sepsis models. 17,18 In addition, EP has neuroprotective effects against paraquat intoxication, 19 kainic acid-induced neuron death in the hippocampus, 20 and spinal cord ischemic injury. 21
Alpha-ketoglutarate (A-KG) is one of the two ketone derivatives of glutaric acid. A-KG is a key intermediate in Kreb’s cycle and acts as the most important nitrogen transporter in metabolic pathways. It has natural and ubiquitous tendency to collect amino group in the body tissue and fluids. The amino group of amino acids is attached to it by transamination. This amino acid has been reported to have a critical role in the cell’s ability to metabolic adaptation to injury. A-KG is transaminated, along with glutamine, to form the excitatory neurotransmitter glutamate. Glutamate is then decarboxylated to the inhibitory neurotransmitter gamma-aminobutyric acid (GABA), which has neuroinhibitory effects and thus protects from excitotoxicity. In addition, A-KG also acts as a precursor to glutathione. 22
Protective efficacy of EP against MPTP-induced PD in mice has been shown earlier. 23,24 However, efficacy of A-KG against PD remains unexplored. The present study addresses the neuroprotective effects of A-KG and EP against MPTP-induced neurotoxicity in mice. The neuroprotective efficacy of both A-KG and EP was compared with the madopar (MD), a commercially available drug for PD.
Materials and methods
Chemicals
MPTP, (A-KG) and EP were purchased from sigma, Aldrich, Co. (St Louis, MO, USA). MD (Piramal Healthcare Limited, Mumbai) was purchased over-the-counter. All other chemicals were from Merck, Mumbai, India.
Animals
Male albino mice (25–30 g) bred in the animal facility of DRDE, Gwalior, were maintained on a bedding of rice husk in polypropylene cages. Animals had access to water and rodent pellet feed ad libitum. All the animals were fasted 2 h prior to the experiment and were given food 30 min after treatment. The study was approved by the establishment’s ethical committee on animal experimentations.
Treatments
A total of 30 male mice were divided into 5 groups of 6 each. Group I (control) received intraperitoneal (ip) treatment of normal saline daily for 10 days. Group II received oral (po) treatment of normal saline daily for 5 days and thereafter was given MPTP free base (20 mg/kg; ip) daily for the remaining 5 days. Groups III, IV and V were administered (po) A-KG (500 mg/kg), EP (100 mg/kg) and MD (5 mg/kg), respectively, for 5 days and thereafter were given MPTP (20 mg/kg; ip) followed by A-KG, EP and MD (po; +10 min), respectively, daily for the remaining 5 days. All the animals were sacrificed on the 11th day by cervical dislocation under ether anesthesia, and the brain was excised quickly on ice.
Motor coordination (rotarod task)
A rotarod (Orchid Scientific, India) with a 3–cm-diameter rod was used. The rod was elevated 20 cm above the floor, and the animals were made to balance at a constant speed of 20 rpm. The time of fall (in seconds) was recorded by a sensor. Each mouse was subjected to three rotarod trials and the mean value was calculated.
Measurement of reduced glutathione (GSH)
Glutathione levels in the tissue were assayed following the method of Hissin and Hilf. 25 GSH standard curve was established using 3, 6, 12, 24, 48 and 96 μg of GSH in phosphate EDTA buffer. For samples, 0.5 ml of metaphosphoric acid (25%) was added to 0.5 ml of brain homogenate (prepared in phosphate EDTA buffer) and centrifuged at 18,000 rpm for 30 min at 4°C. Then, 2.25 ml of phosphate EDTA buffer was added to 0.25 ml of supernatant and 50 μl of this was separated to which 900 μl of phosphate EDTA buffer was added; 50 μl and 100 μl of O-pthaldialdehyde were added to the samples and standards, respectively, mixed well and incubated at room temperature for 15 min. Fluorescence was measured using a spectrofluorophotometer (Biotek USA) at an Ex. wavelength = 350 nm and Em. wavelength = 420 nm, and the values were expressed as micrograms of GSH/mg protein.
Measurement of malondialdehyde
To assess the amount of lipid peroxidation, malondialdehyde (MDA) levels in the tissue were assayed following the method of Okhawa et al. 26 Briefly, 10% of tissue homogenate (in 1.15% KCl) was prepared and centrifuged for 10 min at 6000 rpm. To 0.1 ml of the above supernatant, 0.2 ml of 8.1% SDS was added, followed by the addition of 1.5 ml of 20% acetic acid (pH 3.5) and 1.5 ml of 0.8% thiobarbituric acid (TBA), this was followed by boiling at 95°C for 1 h. The cocktail was cooled and 1 ml of distilled water and 5 ml of the mixture of n-butanol and pyridine (15:1, v/v) were added and shaken vigorously and centrifuged at 6000 rpm for 15 min. The organic layer was taken and its absorbance was measured at 532 nm. As MDA is not stable, 1,1,3,3-tetramethoxypropane (TMP) was used as an external standard for MDA assay. TMP is hydrolyzed during acid incubation step which generates MDA. From the 20 µM stock solution of TMP (freshly prepared) 0.1, 0.2, 0.4, 0.8 and 1.6 µM were taken and treated similarly as that of the samples to establish a standard curve for MDA. The values were expressed as nanomole MDA/mg protein.
Superoxide dismutase assay
The superoxide dismutase (SOD) levels in the brain homogenate were measured by SOD assay kit (Calbiochem, Germany) by following the manufacturer’s protocol. The kit utilizes tetrazolium salt for the detection of superoxide radicals generated by xanthine oxidase and hypoxanthene. One unit of SOD is defined as the amount of enzyme needed to exhibit 50% dismutation of the superoxide radicals. The values were expressed as units of SOD/mg protein.
Measurement of complex I
Complex I (NADH: ubiquinone oxidoreductase, EC 1.6.5.3) is the first complex of the oxidative phosphorylation, which was assayed following the method of Janssen et al. 27 In this assay, 2,6-dichloroindophenol (DCIP) is used as a terminal electron acceptor. Complex I oxidizes NADH, and the electrons produced reduce the artificial substrate decylubiquinone that subsequently delivers the electrons to DCIP. The DCIP is reduced spectrophotometrically at 600 nm. As the electrons produced by other NADH dehydrogenases are not accepted by decylubiquinone, reduction of DCIP is almost completely caused by complex I activity. Values were expressed as mU/mg protein.
Myeloperoxidase assay
The myeloperoxidase (MPO) levels in brain homogenate were measured using a commercial kit (USCN, Life Science Inc. China) by following the manufacturer’s instructions. The kit is sandwich enzyme immunoassay for in vitro quantitative measurement of MPO in mouse plasma. In brief, precoated wells with antibody specific to MPO were used. Standards or samples were then added to the appropriate microtiter plate wells with a biotin-conjugated antibody preparation specific for MPO. Avidin conjugated to horseradish peroxidase (HRP) is added to each microplate well, incubated and tetramethylbenzidine substrate solution was added. The reaction was stopped using sulfuric acid solution. The change in color was measured spectrophotometrically at 450 nm. The values were shown as MPO ng/ml of plasma.
Protein estimation
The total protein concentration of cells was estimated by the Folin-phenol method of Lowry. 28
Histopathology
For histopathological study, the brain was dissected out and fixed in Bouin’s fluid, 4–6 mm thick transverse sections of the brain were processed in alcohol, cleared in toluene using Leica TP 1020 automatic tissue processor and embedded in paraffin wax. The sections of 10 μm thickness were taken using Micron HM 360 automatic microtome. Sections were stained with hemotoxylin and eosin in Leica Autostainer XL. Slides were observed under Leica DMLB bright field microscope and were analyzed using Leica Qwin V3 image analysis software.
Immunohistochemistry
Paraffin-embedded brain sections were deparafinized in xyline and then hydrated in a graded series of ethanol with a final wash in distilled water followed by 20 min incubation in phosphate-buffered solution (PBS; pH 7.4). Quenching was performed for blocking endogenous peroxide in 3% H2O2 methanol bath followed by permeation with 0.3% proteinase K at 37°C for 30 min. The sections were blocked using normal horse serum (vecta stain Elite ABC kit, Burlingame) for 1 h followed by incubation with mouse polyclonal Anti-alpha-Synuclein antibody (Calbiochem), dilution 1:300 and tyrosine hydroxylase antibody (abcam), dilution 1:100 overnight at 4°C in a humidity chamber. Negative controls were also processed by excluding the step of primary antibody on brain section for nullifying nonspecific staining. After overnight incubation with primary antibody at 4°C, the sections were incubated with biotinylated secondary antibody for 1 h followed by incubation with avidin–biotin conjugated horseradish peroxidase streptavidin (Vectastain Elite ABC kit, Burlingame) in PBS for 45 min at room temperature in a humidity chamber. Visualization was accomplished by incubating in diaminobenzidine working solution (Vector) for 5 min at room temperature and counterstaining with diluted methyl green for 30 s followed by rinsing in Scott’s water. Finally, the sections were dehydrated in ethanol, cleaned in xylene and mounted for microscopic observation.
Statistical analysis
The results are expressed as mean SEM (n = 6). The data were analyzed by the one-way analysis of variance followed by Student-Newman-keuls test for comparing control and the various groups, using Sigmastat software (Jandel Scientific Inc., USA). Statistical significance was estimated at 5% level.
Results
Effect of MPTP on motor coordination
There was a significant (p < 0.05) effect of MPTP intoxication in mice on the rotarod performance. The rotarod test (for motor coordination) was performed on a day-to-day basis and significant changes were observed on the fourth and fifth day (Figure 1). A-KG and EP treatment group showed significant improvement in the rotarod performance of the animals and was comparable to that of MD.

Effect of MPTP on motor coordination of mice as measured by rotarod activity. MPTP significantly reduced the motor coordination of mice, which was resolved by treatment with A-KG, EP or MD. Each value is a mean of 3 observations performed on 6 animals each ± SEM. Significance was determined by one-way ANOVA, followed by Student–Newman–Keuls test. *Significantly different from control, #Significantly different from MPTP control (p < 0.05). A-KG: alpha-ketoglutarate; EP: ethyl pyruvate; MD: madopar; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
MPTP-induced oxidative stress
To examine the efficacy of A-KG and EP in curtailing MPTP-induced oxidative stress in mouse brain, GSH, MDA and SOD levels were measured in brain homogenate. The levels of potential antioxidant GSH was significantly lowered by MPTP as compared to control (p < 0.05). Treatments with A-KG and EP significantly attenuated the GSH levels which were comparable to the protection afforded by MD (Figure 2(a)). Corresponding to the reduction in GSH level, significantly elevated levels of MDA were observed in MPTP-treated mice. Treatments with A-KG and EP significantly reduced the levels of MDA which were comparable to MD (Figure 2(b)). MPTP also caused significant inhibition of SOD activity in mouse brain as compared to control (p < 0.05). A-KG but not EP was found to improve the SOD activity, which was at par with that of MD (Figure 2(c)).

Effect of MPTP on the oxidative stress in brain as evidenced by significantly reduced GSH content (a); increased MDA levels (b) and inhibited the SOD activity (c), which were resolved by treatment with A-KG, EP or MD. Values are the mean of 6 animals ± SEM. Significance was determined by one-way ANOVA, followed by Student–Newman–Keuls test. *Significantly different from control, #Significantly different from MPTP control (p < 0.05). MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; GSH: glutathione; MDA: malondialdehyde; SOD: superoxide dismutase; A-KG: alpha-ketoglutarate; EP: ethyl pyruvate; MD: madopar.
Evaluation of neuroinflammation
To investigate the effect of A-KG and EP on MPTP-induced inflammation in mouse brain, the MPO activity was measured. None of the treatments produced significant alterations in the levels of MPO (Data not shown).
Effect of MPTP on complex I activity of electron transport chain
Complex I is a very important enzyme system in the electron transport chain. MPTP caused significant inhibition of the complex I activity after 5 days of treatment. Treatment of both EP and A-KG offered significant protection against the toxic effect of MPTP. The activity of complex I was significantly restored by both the treatments, and the results were comparable with MD (Figure 3).

Effect of MPTP on the complex I activity of the electron transport chain in brain. MPTP significantly inhibited the complex I activity, which was resolved by treatment with A-KG, EP or MD. Values are the mean of six animals ± SEM. Significance was determined by one-way ANOVA, followed by Student–Newman–Keuls test. *Significantly different from control, #Significantly different from MPTP control (p < 0.05). MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; A-KG: alpha-ketoglutarate; EP: ethyl pyruvate; MD: madopar.
Histological investigation
There were no noteworthy changes in the histology of midbrain of mice after 5 days of MPTP treatment as compared to control. However, increased gliosis was observed in the MPTP-treated brain sections which were found to be moderately regressed following A-KG, EP and MD treatments (Figure 4(a) to (e)).
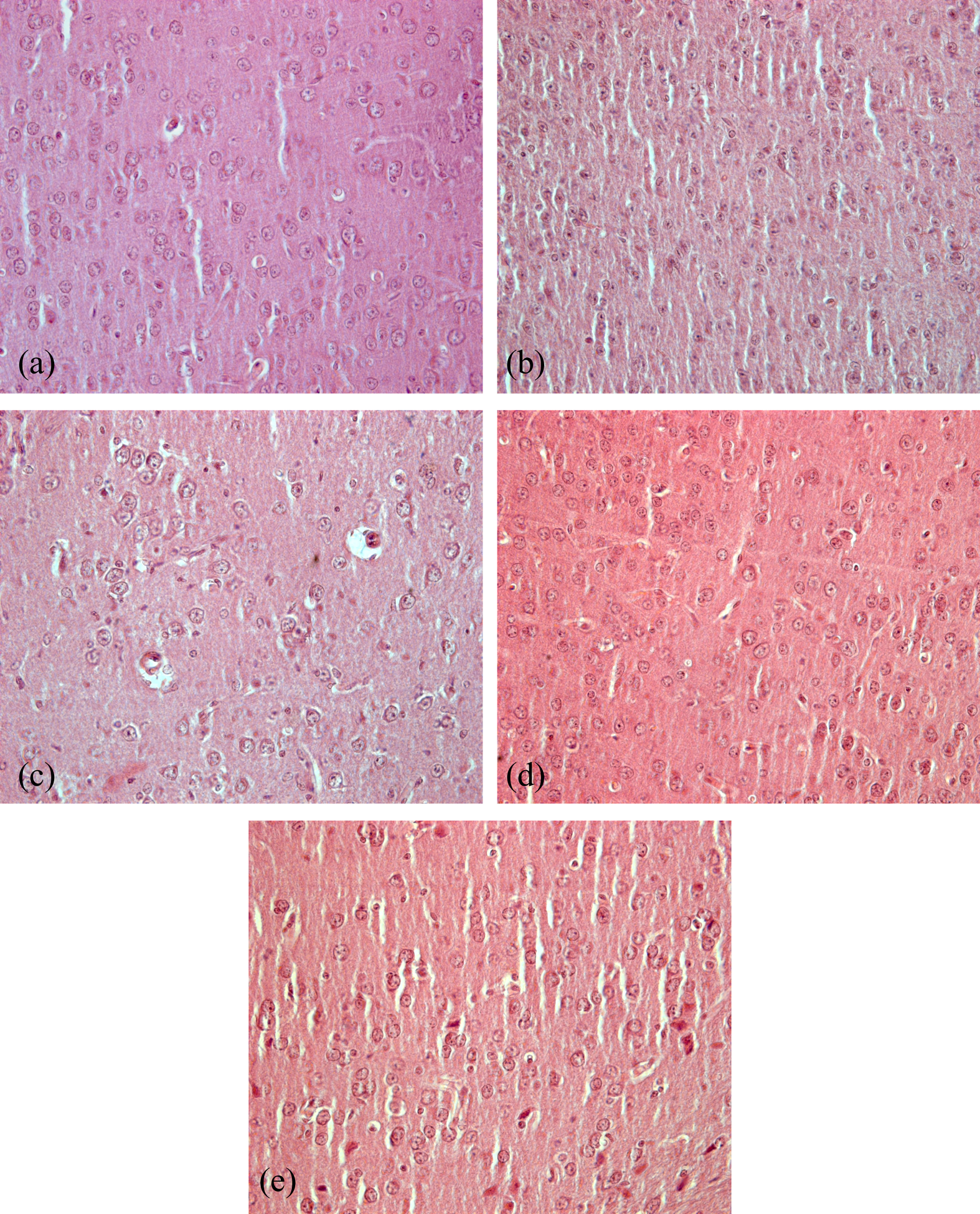
Histograms showing the lateral sections of the midbrain region of mice. As compared to control (a), mild gliosis was observed in MPTP group (b). Protective effects of A-KG (c), EP (d) and MD (e) were observed in MPTP-treated animals (20×). MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; A-KG: alpha-ketoglutarate; EP: ethyl pyruvate; MD: madopar.
Immunohistochemical investigations
Tyrosine hydroxylase (TH) is the rate-limiting enzyme in dopamine biosynthesis. Immunostaining of the midbrain sections using an antityrosine hydroxylase antibody demonstrated A-KG and EP treatment which significantly reduced MPTP-induced TH-positive dopaminergic neuronal loss relative to the MPTP group in the midbrain (Figure 5(a) to —(e)). In control mice, the cytoplasm and fibers of dopaminergic neurons were intensively stained and the cellular processes were evident, showing pronounced immunoreactive positive signals (Figure 5(a)). By contrast, MPTP treatment resulted in a marked loss of dopamine-containing neurons, and few immunoreactive-positive cells were seen (Figure 5(b)). However, in the A-KG- and EP-treated groups, numerous immunoreactive-positive cells were evident. EP treatment demonstrated a significant attenuation of MPTP-induced loss of dopaminergic neurons as compared to A-KG treatment (Figure 5(c) and (d)). Significant protection was also conferred by MD against MPTP-induced toxicity on dopaminergic neurons (Figure 5(e)). To determine whether MPTP-induced oxidative stress causes α-synuclein pathology, the midbrain sections of MPTP-treated mice were examined for α-synuclein-positive cells using immunohistochemical method. In control mouse, relatively faint stained α-synuclein-immunoreactivity was easily identifiable (Figure 6(a)). Dark stained α-synuclein-immunoreactivity was observed in the midbrain sections of MPTP-treated mouse (Figure 6(b)), indicating overexpression of α-synuclein. The treatment with A-KG and EP was found to be effective against MPTP-induced overexpression of α-synuclein. A-KG and EP reduced the staining for α-synuclein (Figure 6(c) and (d)) and was comparable to that of MD (Figure 6(e)).
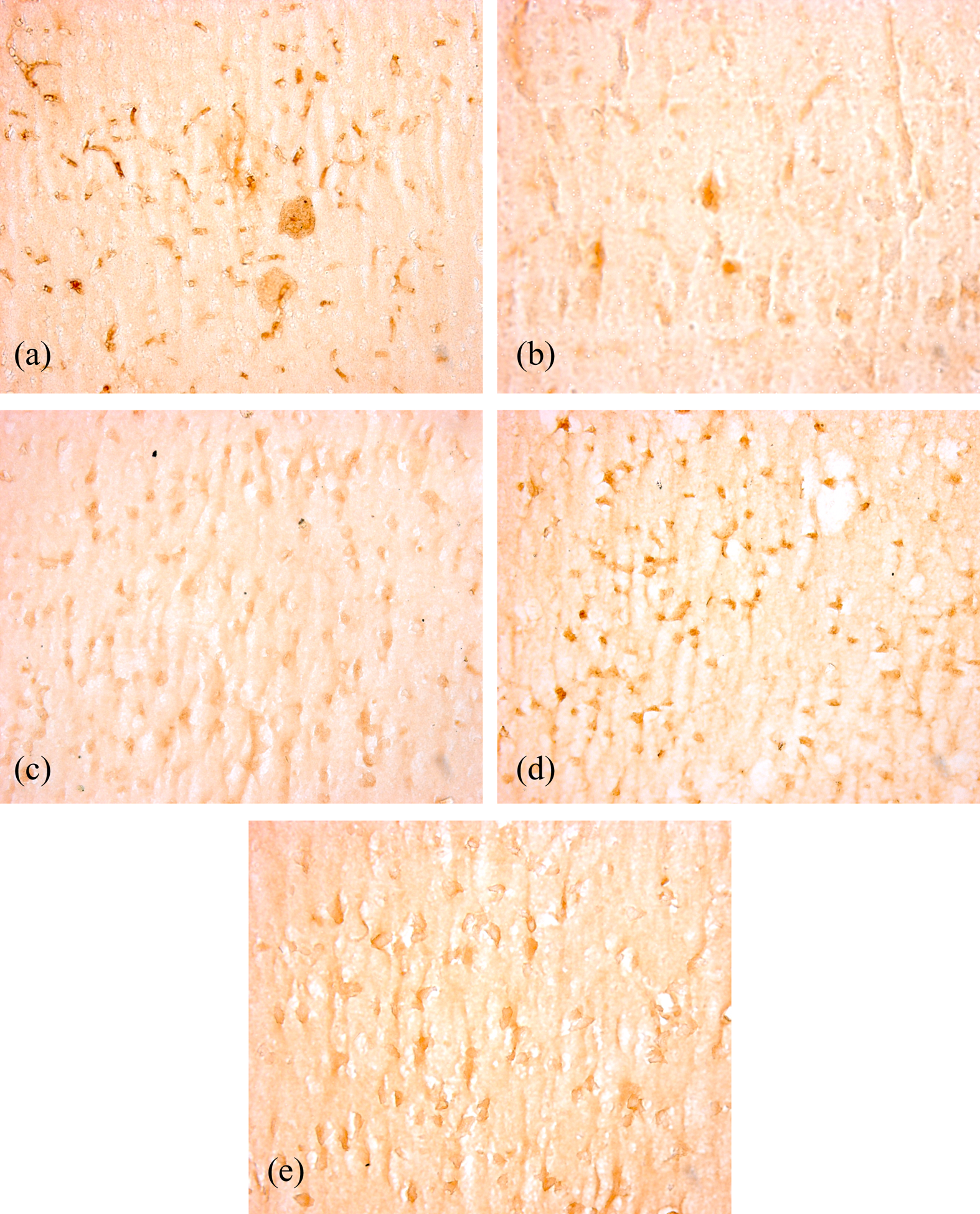
Photomicrographs showing the immunohistochemistry of TH-positive stains in the midbrain region of mice. As compared to control (a), MPTP-treated group (b) showed decrease in the TH-positive cells, which favorably responded to treatments of A-KG (c), EP (d) and MD (e) (20×). TH: tyrosine hydroxylase; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; A-KG: alpha-ketoglutarate; EP: ethyl pyruvate; MD: madopar.

Photomicrographs showing the immunohistochemistry of alpha-synuclein-positive stain in the midbrain region of mice. As compared to control (a), MPTP-treated group (b) showed an increase in the alpha-synuclein-positive cells, which favorably responded to treatments of A-KG (c), EP (d) and MD (e) (20×). MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; A-KG: alpha-ketoglutarate; EP: ethyl pyruvate; MD: madopar.
Discussion
The present findings reveal a novel neuroprotective effect of A-KG in the mouse MPTP model of PD, whereas the effectiveness of EP is relatively known. Treatment with both these carboxylic acids improved the motor behavior and attenuated the oxidative stress caused due to electron transport chain inhibition and improved the histological and biochemical output of MPTP-lesioned mice.
The MPTP model in mice is widely used to study the neuroprotective effect of drugs because it recapitulates the primary pathological and biochemical features of PD, such as oxidative stress and mitochondrial dysfunction. 29 Treatment with A-KG and EP ameliorated the MPTP-induced behavioral deficits. Both these treatments increased spontaneous locomotion and motor coordination relative to MPTP-only treated mice, and these effects were comparable to that of MD.
Reduced activity in mitochondrial complex I (NADH ubiquinone oxidoreductase) is associated with a wide spectrum of neurodegenerative diseases. 30 Reduced complex I activity has also been reported in both autopsy brain tissues and platelets of patients affected with PD. 31,32 MPTP shows considerable inhibition of complex I activity that results in diminished energy metabolism and oxidative stress. The elevated levels of thiobarbituric acid reactive substances (TBARS) in the brain of MPTP-treated mouse observed in the present study suggest that lipid peroxidation is increased after MPTP treatment. The high amount of TBARS was also reported in the brain of MPTP-treated monkeys 33 and is also evidenced in the autopsied brain samples of PD patients. 34 Treatment of MPTP mouse with A-KG and EP reduces the TBARS level which suggests that both these compounds are able to limit lipid peroxidation. Depletion of GSH is the earliest biochemical change in the brains of PD patients. 35 The enzyme SOD is involved in the dismutation of superoxide into oxygen and hydrogen peroxide, while latter is scavenged by glutathione peroxidase and catalase. CNS cells are vulnerable to free radical toxicity because of high rates of catecholamine oxidative metabolic activity. 36 We observed the reduced levels of GSH and SOD in the MPTP-treated mouse midbrain region. Improvement in GSH levels and SOD activity after A-KG and EP treatment observed in the present study suggests that both these compounds show antioxidant properties.
Neuroinflammation and oxidative stress are the two main causes of neuronal cell death in neurodegenerative diseases. Histopathological examination of brains of people suffering from PD generally reveals a moderate to mild gliosis that probably is a consequence of neuronal loss and scarf formation. 37 Inflammation is strongly suggested to contribute to the plaque formation in Alzheimer's disease (AD). As in PD, cytokines, chemokines and complement factor are detected in cerebrospinal fluid and in plaques from patient with AD. 38 In this study, though the MPO level is unchanged, neuronal gliosis was observed in the brain tissue, indicating the involvement of neuroinflammation. A number of in vivo and in vitro studies have demonstrated that EP is an anti-inflammatory agent 20,39 –41 and an effective scavenger of ROS, 42,43 but no improvement in SOD activity in this study does not go well with the available literature. In addition, EP showed neuroprotective effects in the rodent models of stroke 44 and salutary effects on cell cultures and animal models of neurological diseases. 20,39 EP also protected against dopamine-induced apoptosis in PC12 cells. 20,45 Herein, we showed that EP suppressed the TH0positive neuronal cell death in the midbrain region. The degeneration of these neurons is a hallmark of PD in the MPTP-intoxicated mouse models. In addition, EP administration also reduced the accumulation of alpha-synuclein in this region of brain. These findings show that EP plays a neuroprotective role in the MPTP-intoxicated mouse model of PD.
Deamination of glutamine via glutaminase produces glutamate a precursor of GABA, a neurotransmission inhibitor. Glutamate plays an important role in neuronal differentiation, migration and survival in the developing brain via facilitated Ca2+ transport. 46 It also plays a critical role in synaptic maintenance and plasticity. 47 It contributes to learning and memory through use-dependent changes in synaptic efficacy and plays a role in the formation and function of cytoskeleton. Yamamoto and Mohanan 48 investigated the effects of A-KG on kainic acid–induced brain mitochondrial DNA damage and seizures in mice. The increased lipid peroxidation in vivo and in vitro due to kainic acid exposure was completely inhibited by A-KG treatment. They concluded that A-KG would inhibit ROS-dependent oxidative damage to macromolecules in the brain cells. Antioxidative properties of various concentrations of different alpha-ketoacids including A-KG and their effects on the hemolysis of human erythrocytes induced by H2O2 were studied by Sokolowska et al. 49 They concluded that A-KG offers protection against oxidative stress by participating in H2O2 decomposition process. In this study, similar results were observed indicating antioxidant properties of A-KG. It also reduced the cell death due to oxidative stress caused by MPTP treatment in the midbrain region.
Levodopa (
In summary, both A-KG and EP cross the blood–brain barrier, act as antioxidants as well as energy substrates, thereby, immediately improving the energy metabolism of the compromised cells. Further studies are required for the development of these compounds as possible antidotes for MPTP toxicity and PD treatment.
Footnotes
Acknowledgments
The authors thank Prof (Dr) M.P. Kaushik, Director, Defence Research and Development Establishment, Gwalior for his support and guidance.
Conflict of Interest
The authors declared no conflicts of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.