
Abstract
Nitric oxide (NO) is an important physiological signaling molecule. However, when produced in excessive amounts, NO can also have toxic effects. The aim of this study is to investigate the effects of exogenous- and endogenous-derived NO on oxidative modifications of proteins and apoptosis in activated platelets. Washed platelets were incubated with
Introduction
Apoptosis, or programmed cell death, is a main physiologic mechanism that regulates clearance of cells. Over the last decade, it has been discovered that apoptosis occurs not only in nucleated cells but also in anucleated cells such as platelets. 1 Platelets show many of the signs of the apoptotic mechanism such as depolarisation of the mitochondrial membrane potential (ΔΨm), microparticle formation, activation of caspase-3 and exposure of phosphatidylserine (PS) on the platelet surface. 2,3 Apoptosis is frequently accompanied by the generation of reactive oxygen and nitrogen species (RONS). 4 However, a specific role of RONS in apoptotic cell death has not been established yet. Platelets can also generate RONS that may play an important role in platelet functions. But, increased RONS were reported to provoke oxidative reactions in platelets. 5
Nitric oxide (NO) is an important messenger molecule involved in many biological processes. NO is synthesized from
An important feature of NO is that its properties and cellular targets at higher concentrations are profoundly different, particularly under conditions of oxidative stress, where it rapidly reacts with superoxide to form peroxynitrite which is an RONS. 12 Peroxynitrite is a potent oxidant. Peroxynitrite not only decreases NO biovailability but also induces oxidation and/or nitration of cellular components such as DNA, lipids, proteins and low-molecular-weight biomolecules. When peroxynitrite is generated in excess, it may damage cells by oxidizing and nitrating cellular components. Nitration of some amino acid residues, particularly tyrosine in proteins by peroxynitrite can result in the alteration of protein function or structure and inhibition of enzyme activities. Nitrotyrosine (NT) has been used extensively as a marker of peroxynitrite formation. Thiols also are the preferential targets of peroxynitrite. 13 Exposure to NO or activation of iNOS has been reported to inhibit 14 or stimulate 15 apoptosis in several cell types.
It has been observed that incubation of platelets with peroxynitrite nitrated the platelet proteins and impaired platelet functions.
16
But it is still unclear whether
Material and methods
Blood collection
Blood was collected from healthy donors (12 subjects) with a mean age of 22 years. All donors were nonsmokers; none of them have been taking aspirin or other drugs affecting platelet function for at least 7 days prior to blood collection and none of them had a medical history suggesting haemostatic disorders.
Preperation of washed platelets and platelet-rich plasma (PRP)
Blood collected from healthy volunteers was anticoagulated with 1/9 volume of acid citratedextrose (ACD, 2.5% trisodium citrate, 2.0%
For aggregation experiments, fresh blood was anticoagulated with 1/9 volume of 3.8% trisodium citrate and centrifuged at 150g for 10 min at room temperature to get PRP.
Incubations
For protein modification and apoptosis experiments, washed platelets were suspended in the modified Tyrode’s buffer (127 mM NaCl, 2.7 mM KCl, 0.5 mM NaH2PO4, 12 mM NaHCO3, 5 mM HEPES ve 5.6 mM glucose, 1 mM MgCl2 and 1 mM CaCl2, pH: 7.4). Adenosine diphosphate (ADP) (20 µM) was used to trigger oxidative stress and apoptosis in the platelets. Platelet suspensions were incubated with
Mitochondrial membrane potential (ΔΨm), PS exposure and P-selectin levels were analyzed by flow cytometry as mentioned below.
NO assay
The NO levels in platelets were measured using the method of Chen et al.
18
with minor modifications, indicated as nitrite plus nitrate (NOx) which is commonly used to monitor NO concentration. 1 mM
Platelet aggregation
To assess the effects of exogenous
Assay of oxidative/nitrosative stress parameters
Protein carbonyl (PCO) and NT formation were measured as indices of oxidative/nitrosative stress. Carbonyl groups (aldehydes and ketones) are produced on protein side chains (especially of proline, arginine, lysine and threonine) when they are oxidized. These moieties are chemically stable. 13 The contents of PCO were assayed according to the method of Levine et al. 19 with minor modifications. Supernatants were incubated with 10 mM 2,4-dinitrophenylhydrazine (DNPH) in 2 N HCl for 1 h at room temperature. Protein hydrazone derivates were precipitated with TCA 20% and then the precipitates were washed three times with ethanol:ethyl-acetate (1:1). The final pellet was resuspended in 6 M guanidine hydrocholoride and incubated for 15 min at 37°C. The absorbance was measured at 360 nm, using a molar extinction coefficient of 2.2 × 104 M−1 cm−1. The results were expressed as nmol PCO/mg protein.
NT modification of proteins is a well-established marker of protein damage by oxidative stress. 3-nitrotyrosine is a product of protein tyrosine nitration resulting from oxidative damage to proteins by peroxynitrite. 13 The levels of NT in platelet proteins were measured by Nitrotyrosine Chemiluminescence Detection Assay Kit following the protocol supplied by the manufacturer (Millipore, Cat No: 17-376). Briefly, 96-well plates were coated with 5 μg/mL nitrated BSA overnight at 4°C, and blocked with blocking buffer for 1 h; 50 μL of sample or standard and 50 μL of 2X anti-NT were added to each well, then the plates were incubated at 37°C for 60 min; 100 μl per well of 1X anti-rabbit immunoglobulin G (IgG), horse radish peroxidase (HRP) conjugate was added and incubated at 37°C for 60 min. The freshly prepared LumiGLO® Chemiluminescent Substrate (75 μL) was added to wells and incubated at room temperature for 10 min. The plates were washed between each step. The amount of NT was calculated with the standard curve and expressed as µg nitrated BSA equivalents per mg protein.
Caspase-3 activity assay
Caspase activity was measured using a caspase-3 colorimetric assay kit (R&D Systems BF3100) following the manufacturer’s protocol. This method uses a colorimetric assay to monitor cleavage of an acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) substrate, which resembles the caspase-3 cleavage site. First, platelets were lysed in the lysis buffer at room temperature for 15 min. The platelet lysates were then centrifuged at 2000g for 10 min at 4°C to precipitate cellular debris. Cell lysates were incubated with substrate solution Ac-DEVD-pNA for 1 h at 37°C. In order to measure specific caspase-3 activity, platelets were also treated with a caspase-3-specific inhibitor, N-Ac-Asp-Glu-Val-Asp-CHO (Ac-DEVD-CHO, Sigma A0835). The absorbance was read at 405 nm using a micro reader. The caspase-3 activity was calculated from the cleavage of the specific colorimetric substrate (Ac-DEVD-pNA). The difference between the amount of pNA produced in the absence of inhibitor and in the presence of inhibitor is a measure of specific caspase-3 activity. The results were expressed as nmol pNA/mg protein.
P-Selectin expression assay
For determination of platelet activation, P-selectin expression was monitered as described previously.
17
Platelets were treated with ADP, ADP +
PS exposure assay
For PS exposure experiments, platelet suspensions of 100 μL aliquots were placed in siliconized glass cuvettes at 37°C. ADP,
ΔΨm assay
Changes in ΔΨm were measured according to the kit procedure (MitoPT JC-1 #911) using the J-aggregate-forming lipophilic cationic fluorochrome 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol carbocyanine iodide (JC- 1). Briefly, JC-1 was dissolved in 500 μL dimethyl sulfoxide (DMSO) for preparing the stock solution and then diluted in platelet buffer on the day of staining. First, washed platelets (5 × 108 /ml) were incubated with agents as described above at 37°C for 30 min. Then, 5 μL JC-1 was added to tubes and incubated for 20 min in the dark at 37°C. Platelet buffer (1.5 ml) was added and centrifuged at 1000g for 5 min and the supernatant was discharged. Buffer was added and centrifuged at 1000g and the supernatant was removed. Buffer (1 ml) was added and samples were analyzed immediately by flow cytometry.
For flow cytometric analyses, all the samples which were prepared as described above were analyzed on a Beckman Coulter, EPICS XL-MCL flow cytometer or a Becton-Dickinson, FACScan. The flow cytometer was equipped with a 488 nm argon ion laser. The platelet population was identified by forward scatter for cell size and by side scatter for cell granularity. Alignment of the instrument was checked daily by calibration beads. An electronic bitmap was placed around the platelet population and CD41a-FITC was used to form a gate. The results were expressed as the percentage of antibody-positive platelets. Nonspecific and background fluorescence was determined by the use of FITC-conjugated IgG. Fifty thousand platelets were counted in each tube.
Protein assay
Protein content was analyzed according to Bradford’s method 20 using bovine serum albumine as the standard and expressed as milligram protein per milliliter platelet suspension.
Statistical analysis
Data are presented as mean ± standard deviation (SD). Statistical analysis was carried out using the Instat statistical package (GraphPad Software, San Diego, California, USA). The statistical difference between groups was determined by the Student's t test. A p value less than 0.05 was considered statistically significant.
Results
NO production
Washed platelets in the presence of ADP were incubated with or without

Mean values of NOx levels in platelets (+++ p < 0.001 compared to baseline, **p < 0.01 compared to ADP). NOx: nitrite plus nitrate; ADP: adenosine diphosphate.
Oxidative/nitrosative stress parameters in platelet protein
We measured the levels of PCO and NT as markers of protein modification. Figure 2(a) shows the levels of PCO after ADP and ADP+

Mean values of PCO (a) and NT (b) levels in platelets (+ p > 0.05 compared to baseline, *p < 0.05, **p < 0.01 compared to ADP). PCO: protein carbonylation; NT: nitrotyrosine; ADP: adenosine diphosphate; GSNO: nitroso-glutathione.
NT levels of platelet proteins are shown in Figure 2(b). After activation with ADP, there was a significant increase in the NT levels as compared with the baseline group (p < 0.05). Addition of
Platelet aggregation and P-selectin expression
Platelet aggregation responses after

(a) Platelet aggregation responses (%) and (b) a sample of aggregation curve (***p < 0.001 compared to ADP group). ADP: adenosine diphosphate; GSNO: nitroso-glutathione.
Figure 4(a-d) shows the expression of platelet P-selectin as platelet activation marker in all groups. After ADP, there was a significant difference in the expression of P-selectin on the platelet surface as compared with the baseline group (p < 0.001). The expression of platelet P-selectin significantly reduced after
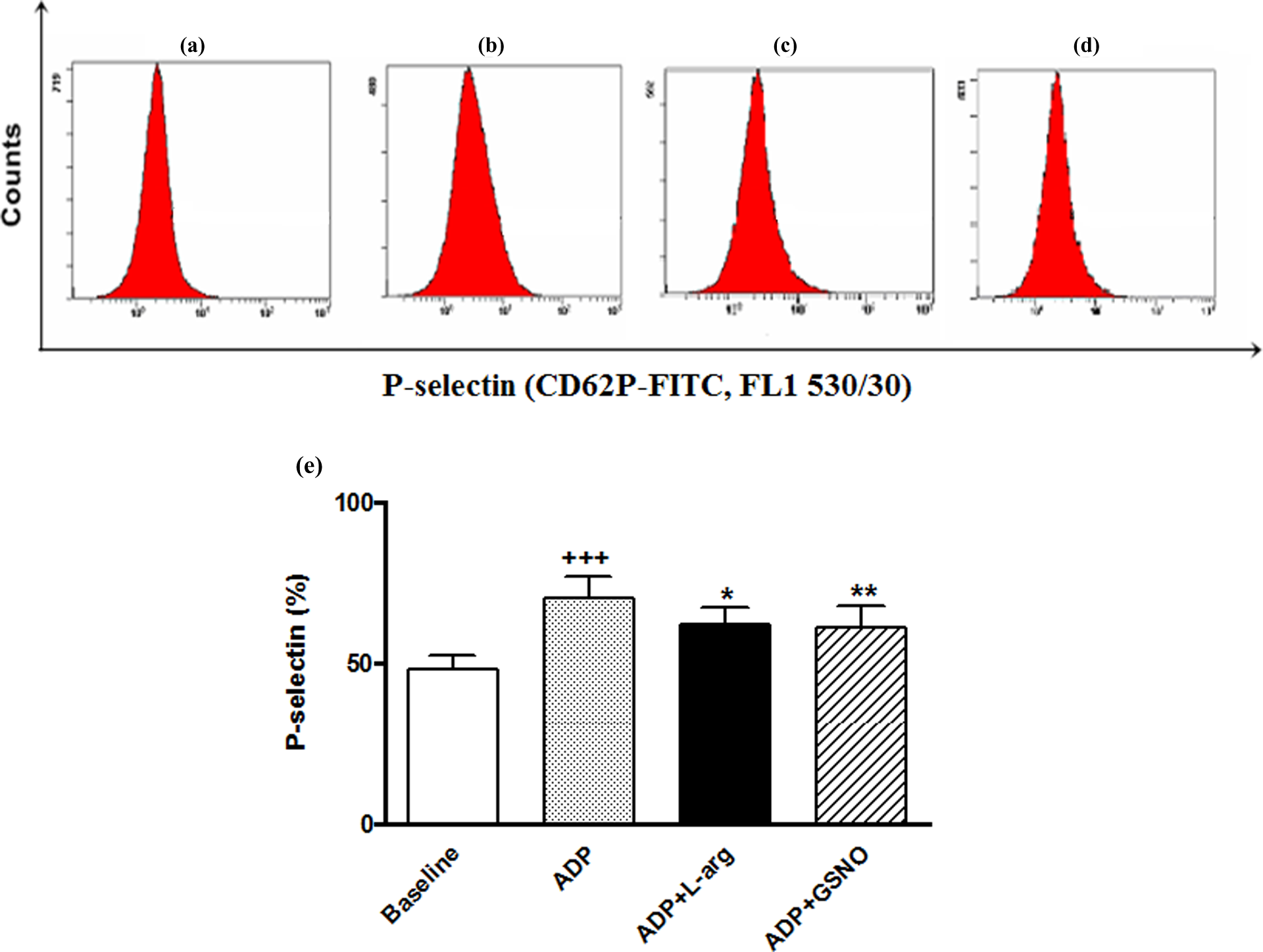
Histogram samples of baseline, ADP, ADP+
Apoptotic events
In order to investigate whether NO can induce platelet apoptosis, platelets were incubated with
Figure 5(a-d) shows flow cytometer histograms of annexin-V binding in platelets.

PS exposure of platelets was determined using annexin-V. (a-d) Flow cytometric histogram samples of baseline, ADP, ADP+
After ADP stimulation of platelets, there was a significant increase in PS exposure as compared with the baseline group (p < 0.001). Treatment with
Caspase-3 is the major effector caspase involved in apoptotic pathways.
1
Caspase-3 levels are shown in Figure 5(f). Activation with ADP caused a significant increase in platelet caspase-3 activity (p < .001). After
To investigate whether the activation of caspase-3 and PS exposure are apoptotic events we have tested using ΔΨm. Cell-permeable lipophilic cationic dye JC-1 was used for the determination of ΔΨm. If the ΔΨm is maintained, this dye accumulates in the mitochondria and shows a shift in fluorescence from green (monomeric form, FL1) to red (aggregate form FL2). Mitochondria have a high ΔΨm, breakdown of ΔΨm is characteristic of apoptosis. 2 In this study, changes of ΔΨm in platelets were monitored by red fluorescence (FL2) in JC-1-stained platelets. Besides, the results were also expressed as dividing the arithmetic mean fluorescence intensity (MFI) of FL2 by the MFI of FL1 (FL2/FL1 ratio).
Figure 6(a-d) shows dot graphs of experimental groups after platelets were stained with JC-1. As shown in Figure 6(E), after platelet activation with ADP, ΔΨm decreased as compared with the baseline group (p < 0.001).

Effects of
Discussion
In this study the role of NO and protein modifications in mechanisms underlying platelet apoptosis which has a crucial importance in thrombosis and hemostasis were investigated. Stimulation of platelets by ADP caused an increase in platelet NO production as observed in previous studies.
18
The intracellular uptake of
The stimulation of the platelets with agonists leads to increases in levels of reactive oxygen species such as superoxide and H2O2; thus, increases the oxidative markers. 5 ADP also causes superoxide production. 22 NO reacts with superoxide at a high rate to form peroxynitrite, which can induce nitration of proteins. 16 The stimulation of platelets results in the activation of several key tyrosine kinases and phosphatases, leading to the phosphorylation of several key cellular proteins. 23 Peroxynitrite-induced nitration of proteins has been shown to reduce their phosphorylation when exposed to specific protein kinases, suggesting that protein nitration may interfere with protein phosphorylation signaling pathways. 16 Exogenous peroxynitrite can activate or inhibit platelets depending on its concentration. 13 In our study, ADP increased carbonylated and tyrosyl nitrated proteins, when compared with resting platelets. During collagen-induced activation of platelets, spontaneous nitration of platelet proteins have been reported. 24 NO is required for nitration of proteins. It was shown that when the platelets are activated with agonists, the expression of iNOS and particularly eNOS increase. In our study, due to the short incubation period the increase in nitrated protein levels could have resulted from the increased activation of the present active iNOS form or eNOS rather than the increase in protein expression of iNOS. 25 In parallel to our findings, small amounts of nitrated proteins were found in resting platelets, while the nitrated protein levels were found to increase as a result of collagen stimulation. 23,24
As the importance of NO has been understood, some several NO releasing agents have gained extensive medical use. S-nitrosothiols are not being used clinically at present, but there are a large number of animal and clinical studies demonstrating their beneficial features. Some of these agents show tissue selectivity; such as S-nitrosoglutathione which is selective for arteries over veins, thus, has a distinct hemodynamic profile of action when compared with the classical organic nitrates. 12 GSNO has been shown to decrease the prevalence of cerebral embolism after carotid endarterectomy in patients already receiving aspirin and heparin. Additionally, GSNO reduces platelet adhesion in bypass grafts. 12 Therefore, we used GSNO as the exogenous NO donor in our study. NO acts upon being released from GSNO. The enzymatic activity necessary for this relase is also present in the platelets besides many other cells. 26,27
We observed that
GSNO caused significant increases in nitrated protein levels, while decreasing the oxidative changes. GSH found in the structure of GSNO may play role in its antioxidative effect. 12 It has been shown that GSNO might lead to NT formation in the vessel wall. 29 We observed that 100 µM GSNO significantly decreased platelet aggregation and activation. These findings are parallel with those of previous studies. 30,31
NO’s effect on cell apoptosis varies depending on NO concentration and type of the cell involved.
32
Apoptosis is frequently accompanied by increasing oxidative stress.
4
The role of increased oxidative stress in NO-induced apoptosis is indicated by the suppression of NO-induced apoptosis by antioxidants.
33
The recent studies verified that platelet apoptosis is induced by physiological agonists or calcium ionophores or platelet storage.
1
In this report, we observed that platelet stimulation with 20 µM ADP resulted in caspase-3 activation, PS externalization and loss of mitochondrial potential. Despite ADP is a weak platelet agonist, it was shown that it caused increases in some platelet apoptotic markers at the concentration we used in our study.
34
At a study conducted on rats, it was shown that oral administration of
It can be concluded that both endogenous NO and exogenous NO provided from GSNO protect the platelets from apoptosis. The protein nitration in platelets is NO- and NO-production-dependent but is independent of increased oxidative stress. This in vitro study can be a basis for novel in vivo studies that would investigate the use of
Footnotes
Conflict of interest
The authors declared no conflicts of interest.
Funding
The research was supported by Marmara University Research Unit Grants SAG-C-YLP-120309-0035.