
Abstract
Low level, antenatal exposure to dioxins is associated with low birth weight, which in turn is associated with long-term sequelae. We exposed the human extravillous cytotrophoblast (EVT) lines HTR-8/SV40 and TCL1 to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and assessed cell growth, invasion, and differentiation. TCDD had no effect on cell proliferation, invasion, or tube formation in Matrigel. The EVT-derived cells expressed a functional aryl hydrocarbon receptor protein; however, TCDD exposure did not alter expression levels of proteins involved in EVT differentiation in early pregnancy, including hypoxia-inducible factor 1A (HIF1A), vascular endothelial growth factor (VEGF), Integrin A1, A6, and AVB3. These results suggest that the reduction in fetal weight induced by dioxin is not the result of vascular remodeling via EVT dysfunction.
Keywords
Introduction
Several studies have associated antenatal low-level dioxin exposure with low birth weight.1–4 Low birth weight in turn increases the risk of developmental delay and learning disabilities, 5 high blood pressure 6 and cardiovascular disease.7,8 Thus, it is important to determine the mechanism by which dioxins affect birth weight as an initial step in managing the effects of dioxin exposure in pregnancy.
The biochemical and toxic effects of the most potent dioxin, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), are mediated by its binding to the aryl hydrocarbon receptor (AhR). The AhR mediates the induction of cytochrome P450 (CYP) enzymes. Induction of these CYP enzymes in turn causes leakage of oxidants, thereby promoting oxidative DNA damage. 9 In utero exposure to TCDD markedly suppresses the development of sinusoids and trophoblast cells and induces apoptosis of trophoblast cells with a concomitant increase in the incidence of fetal death in the setting of hypoxia. Cross talk between the hypoxia-inducible factor (HIF)-mediated pathway and the AhR-mediated pathway plays an important role in this physiological process. 10 One HIF member, HIF1A, also plays a key role in the regulation of extravillous cytotrophoblast (EVT) differentiation.11–14 It is therefore plausible that TCDD, by increasing Reactive Oxygen Species (ROS), may directly affect EVT function during placental development and may cause fetal growth restriction via placental dysfunction. There is little evidence, however, that TCDD affects the biological behavior of trophoblast cells.9,10 In this study, we transiently exposed two human EVT cell lines to TCDD to investigate the effects of TCDD on cell growth, invasion, and differentiation.
Materials and methods
Reagents
TCDD was purchased from Sigma Chemical Co. (St Louis, MO, USA). An HIF1A antibody was purchased from BD Transduction Laboratories (Bedford, MA, USA). The vascular endothelial growth factor (VEGF) antibody was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The AhR antibody was purchased from Thermo Scientific (Rodford, IL, USA). The CYP1A1 antibody was purchased from Enzo Life Science Inc. (Plymouth, PA, USA). The aryl hydrocarbon receptor nuclear translocator (ARNT) antibody was purchased from Affinity BioReagents, (Golden, CO, USA). The Integrin A1, A6, AVB3 antibodies were purchased from Chemicon International Inc. (Temecula, CA, USA). Fluorescein isothiocyanate (FITC)-conjugated anti-mouse immunoglobulin G ([IgG] Molecular Probes Inc, Eugene, OR, USA) was used as a secondary antibody.
Cell lines and cell culture
TCL1 cells were established from mixed primary cultures of cells isolated from chorionic membranes obtained at the time of elective, preterm caesarean sections.11–15 The HTR-8Svneo first-trimester cytotrophoblast cell line, established as described previously, 16 was cultured in RPMI1640 (Nipro, Tokyo, Japan) supplemented with 10% fetal calf serum (FCS, Gibco Invitrogen) or conditioned serum in a humidified atmosphere containing 95% air and 5% CO2 at 37°C.
WST assay
Cellular proliferation was measured both by direct counting and with the WST assay. The WST assay was performed using a commercially available kit (Premix WST-1 Cell Proliferation Assay System, Takara, Shiga, Japan) according to the manufacturer’s protocol. In brief, 5 × 103 TCL1 cells were seeded in a 96-well microplate and exposed to TCDD/dimethyl sulfoxide (DMSO) for 24 hours. Cells were then incubated in the presence of the WST reagent for 2 hours, and the absorbance at 450 nm was measured and used to estimate the cell count.
Detection of apoptosis
Apoptosis was confirmed by a terminal deoxynucleotidyl transferase (TdT)-mediated digoxigenin uridine triphosphate (dUTP) nick-end labeling (TUNEL) assay using a commercially available kit (TUNEL Label Mix; Roche Diagnostics, Tokyo Japan), according to the manufacturer’s protocol. Cells were viewed (magnification ×40 to ×400) and photographed using a microscope, BZ-8100 (KEYENCE, Tokyo, Japan). The number of apoptotic cells was measured by counting the number of TUNEL-positive cells. At least three fields per well were examined; each experimental condition was tested in triplicate.
In vitro migration assay
A quantitative measure of the in vitro invasive ability of TCL1 cells was obtained in a modified Boyden Chamber assay (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s protocol. Briefly, after pretreatment with various agents, a 0.5 mL suspension of TCL1 in serum-free media was added to the upper compartment of the Boyden chamber at a density of 2 × 105 cells/well and incubated for 24 hours at 37°C, with 10% fetal bovine serum (FBS)-supplemented media in the lower compartment. Nonmigrating cells were removed with a cotton swab and the remaining cells were fixed and stained (Diff-Quick Stain Set, Dade Behring, Inc., Newark, DE, USA). Filters were removed from the chamber and mounted for visualization under a Nikon Eclipse TS100 (Nikon, Tokyo, Japan). The number of cells migrating to the lower side of the filter was determined by counting all invading cells in each membrane.
Cell scratch migration assay
Cells were plated in 24-well plates and allowed to form a confluent monolayer. The cell surface was then scratched with a 200 μL pipette tip. Cells were photographed after 6 and 12 hours of incubation, BZ-8100 (KEYENCE, Tokyo, Japan). Cell motility was quantified with National Institutes of Health (NIH) image by measuring the area between the migrating cell boundaries in a marked high-power field. Each experimental condition was tested in triplicate.
Tube-like formation assay
Growth factor-reduced Matrigel (BD Bioscience, Franklin Lakes, NJ, USA) was added (300 μL) to each well of a 24-well plate and allowed to polymerize for 1 hour at 37°C. After pretreatment with various agents for 2 hours, 2 × 105 TCL1 cells were seeded. Cells were incubated at 37°C at room air, viewed (magnification ×40–×400), and photographed using an Olympus IX71 microscope (Olympus, Tokyo, Japan). Tube-like formation was quantitated by counting the number of tube-like structures formed by the connected capillary bridge. 15 At least three fields per well were examined; each experimental condition was tested in triplicate.
Western blotting
Cells were lysed with lysis buffer containing 62.5 mM Tris-HCl (pH 6.8), 100 mM dithiothreitol, 2%(w/v) sodium dodecyl sulfate (SDS), and 10% glycerol. Cellular proteins were electrophoresed in an SDS gel together with a prestained molecular weight marker (BioRad Laboratories, Hercules, CA, USA), transferred onto Immobilon-P (Millipore, Bedford, MA, USA), and analyzed by an immunoblotting system (GE Healthcare Japan, Tokyo, Japan).
Results
HTR-8/SV40 and TCL1 cells express the functional AhR
To address whether HTR-8/SV40 and TCL1 cells express the functional TCDD receptor, we performed an immunoblot analysis for the AhR and CYP1A1 (Figure 1A). Both cell types showed AhR protein expression. After incubation with 1 nM of TCDD, CYP1A1 was induced consistent with a functional AhR.
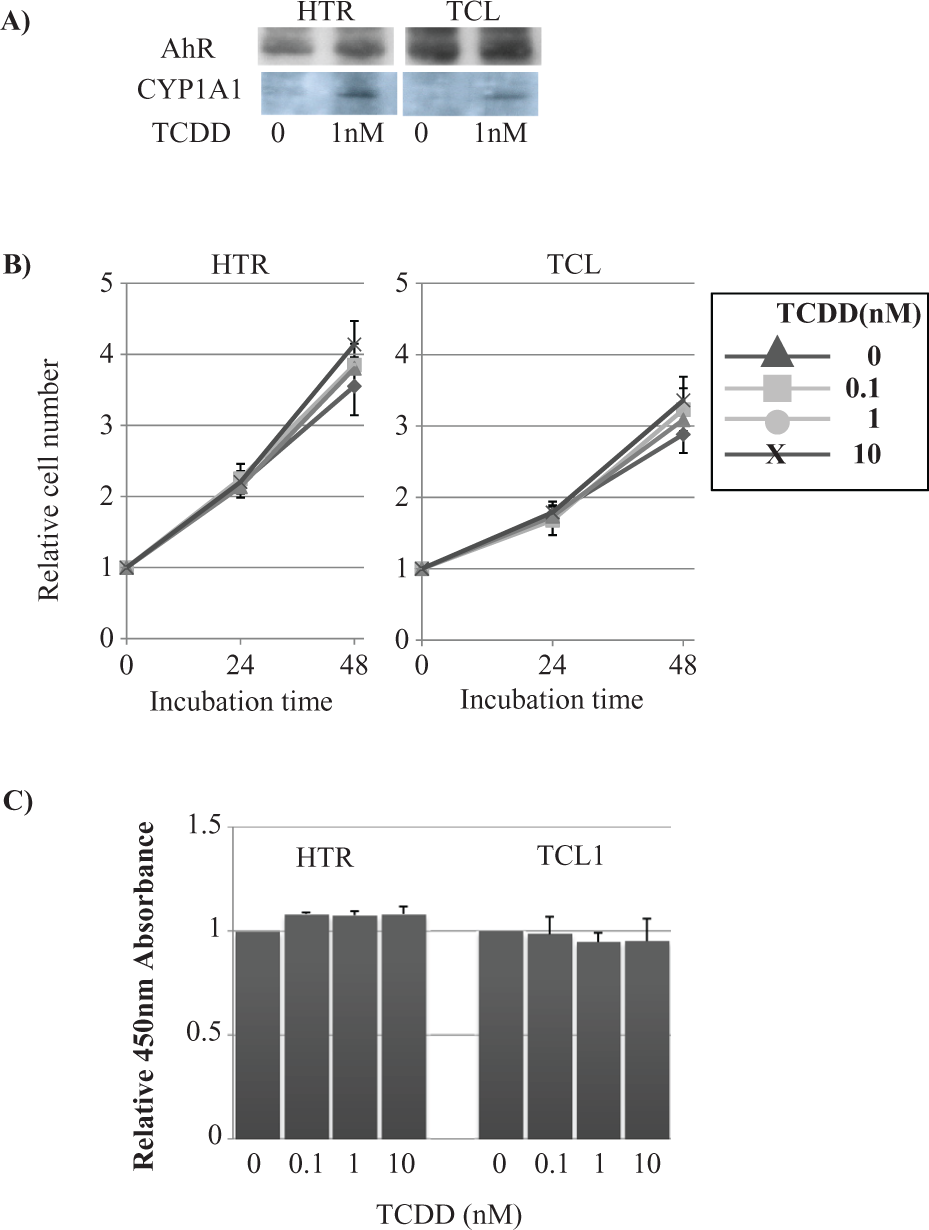
Cellular proliferative activity. (A) Asynchronously growing HTR and TCL1 cells were incubated with 1 nM/2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) for 24 hours. Cellular proteins were then extracted, electrophoresed, and transferred onto Immobilon-P membranes before analysis by immunoblotting using the indicated antibodies. (B) Asynchronously growing TCL1 cells were seeded at 2 × 105/100 mm dish and grown at 37°C. After 1 day of incubation, the medium was replaced with complete medium containing 10% fetal bovine serum (FBS) and the indicated concentration of TCDD. At the indicated time, cells were harvested and both the adherent and nonadherent cells were counted with a Coulter counter. Triangle: 0, Box: 0.1, Circle: 1, and X: 10 nM of TCDD. (C) Asynchronously growing cells were seeded in a 96-well microplate and treated with TCDD/DMSO for 24 hours. A WST assay was then performed using a commercially available kit. Data are represented as the mean ± SD of three independent trials.
TCDD did not affect cellular proliferation and viability in HTR-8/SV40 or TCL1 cells
To examine the effect of TCDD on proliferative activity, we measured cellular growth. After either a 24- or 48-hour incubation with 0.1, 1, or 10 nM of TCDD, there was no significant alteration in cellular growth of TCL or HTR cells (Figure 1B). The relative number of HTR-8/SV40 cells following incubation with 0, 0.1, 1, or 10 nM of TCDD for 24 hours were 2.17 ± 0.19, 2.25 ± 0.38, 2.14 ± 0.11, or 2.20 ± 0.11, respectively. At 48 hours of incubation, there were 3.55 ± 0.41, 3.85 ± 0.23, 3.80 ± 0.15, or 4.14 ± 0.26 cells, respectively (Figure 1A, Left column). The relative numbers of cells following incubation with 0, 0.1, 1, or 10 nM of TCDD for 24 hours were 1.76 ± 0.15, 1.68 ± 0.21, 1.73 ± 0.09, or 1.79 ± 0.11, respectively. At 48 hours of incubation, there were 1.30 ± 0.15, 1.21 ± 0.14, or 0.93 ± 0.13 cells, respectively. At 24 hours, there were 2.88 ± 0.33, 3.23 ± 0.30, 3.09 ± 0.12, or 3.36 ± 0.26, respectively (Figure 1B, Right column).
A WST assay was then performed following a 24-hour incubation with 0.1, 1, or 10 nM of TCDD in these cells. The relative absorbance value at 450 nM after 24 hours of incubation with 0.1, 1, or 10 nM of TCDD compared to the control was 1.08 ± 0.01, 1.08 ± 0.02, or 1.08 ± 0.04, respectively, in HTR cells and 1.47 ± 0.13 and 0.98 ± 0.08, 0.95 ± 0.04, or 0.95 ± 0.11, respectively, in TCL1 cells (Figure 1C). We also performed a TUNEL assay in TCL1 and HTR-8/SV40 cells after a 24-hour incubation with 0.1, 1, or 10 nM of TCDD; however, there was no significant difference in the number of apoptotic cells (data not shown).
TCDD did not affect cellular invasive activities in HTR-8/SV40 or TCL1 cells
To examine the effect of TCDD on the invasion ability of TCL1 and HTR-Neo, we performed a pore membrane motility assay using the modified Boyden-chamber method. The number of invading cells after a 24-hour incubation with 0.1, 1, or 10 nM TCDD was 40.7 ± 4.0, 42.0 ± 6.1, 39.7 ± 8.5, or 42.0 ± 4.6, respectively, in HTR cells (Figure 2A, left column) and 48.7 ± 5.5, 46.7 ± 6.1, 48.7 ± 4.5, or 47.3 ± 6.5, respectively, in TCL1 cells (Figure 2A, right column).

Cellular invasive activity. (A) The number of mi-grating cells was counted in each Boyden chamber membrane. Data are the mean ± SD of three independent trials. (B) A cell monolayer was scratched using a 200-μL pipette tip. Cells were photographed after 6 and 12 hours. Cell motility was quantified by measuring the area between the migrating cell boundaries in marked high-power fields with National Institutes of Health image. Data are the mean ± SD of three independent trials. Triangle: 0, Box: 0.1, Circle: 1, and X: 10 nM of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD).
We then performed a cell scratch test using HTR-8/SV40 and TCL1 cells. The relative split area compared to time 0 after a 12-hour incubation with 0, 0.1, 1, or 10 nM of TCDD was 0.11 ± 0.08, 0.13 ± 0.10, 0.18 ± 0.07, and 0.16 ± 0.02, respectively, in HTR-neo cells (Figure 2B, Left column) and 0.08 ± 0.06, 0.17 ± 0.09, 0.10 ± 0.02, and 0.12 ± 0.07, respectively, in TCL1 cells (Figure 2B, Right column). As shown in Figure 3A, there was no dose-dependent effect of TCDD exposure on cell migration.

Tube formation activity in TCL1 cells. (A) Chronological morphology during tube formation by TCL1 cells after 0 (left) and 12 hours (right) seeded on polymerized Matrigel. Bars: 50 μm. Capillary networks (arrow). (B) The number of capillary networks (arrow in A) per 1-mm2 surface area was counted at magnification ×400. Data are represented as the mean + SD in three independent trials.
TCDD did not affect tube-formation activities in TCL1
Following 12 hours of incubation on Matrigel, TCL1 cells exhibited a morphological change that mimicked endothelial cells termed tube-like formation (Figure 3A). After incubation with 0, 0.1, 1, or 10 nM of TCDD for 8 hours, the number of capillary networks (arrow) was 14.7 ± 2.5, 14.0 ± 4.6, 15.7 ± 3.5, or 14.3 ± 3.1, respectively (Figure 3B).
TCDD did not affect the expression of proteins related to EVT differentiation
We then performed a Western blot analysis to examine the expression of HIF1A, VEGF, ITGA1, A6, AVB3, which are associated with EVT differentiation. Following incubation with 0, 0.1, 1, or 10 nM of TCDD for 24 or 48 hours, there was no significant change in the expression levels of these proteins (Figure 4).

Immunoblotting analysis. Asynchronously growing cells were incubated with 1 nM/2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) for 24 or 48 hours. Cellular proteins were then extracted, electrophoresed, and transferred onto Immobilon-P before analysis by immunoblotting using the indicated antibodies.
Discussion
It is plausible that TCDD affects fetal growth through a variety of mechanisms including directly, following crossing the placenta, and indirectly through disruption of placental development and or endocrine function. Short- and long-term exposure to TCDD may produce different effects. Our study did not demonstrate an effect of short-term TCDD exposure on EVT growth, differentiation, or function.
EVT differentiation has two modes: one is endovascular differentiation, in which EVT replaces and remodels spiral arteries resulting in increased blood flow toward the intervillous space. The other is interstitial invasion, which promotes placental anchorage and is accompanied by endovascular invasion.17,18 Each type is characterized by its own expression pattern of differentiation markers. These gene expression alterations are essential for organization of the fetomaternal interface during early development. For example, integrin subunit conversion during interstitial and endovascular differentiation is well characterized,19,20 and this conversion is defective in patients with placental insufficiency.
In the rat, placental vascular remodeling occurs late in gestation and is characterized by changes in the trophoblast shape and the elimination of trophoblasts by apoptosis. 10 As a result, the net volumes of both the maternal and fetal blood in the placenta increase to meet the requirements for oxygen and nutrients in late gestation. In utero exposure to TCDD markedly suppresses these placental changes and rendered the fetus more susceptible to death from hypoxia. No changes were observed in the cross talk between the HIF-mediated pathway and the AhR-mediated pathway in rats. 10
Controversy exists as to whether TCDD directly affects trophoblastic function as there are few studies investigating this.21–24 TCDD has been shown to significantly increase apoptosis in JAR cells in a dose-dependent manner. 21 Neither TCDD nor a polychlorinated dibenzo-p-dioxin/polychlorinated dibenzo-p-furan (PCDD/PCDF) mixture, however, induces DNA damage or apoptosis in the human placental choriocarcinoma JEG-3 cell line. 22 Additionally, while TCDD has been shown to have differential effects on early, late gestational, and term trophoblasts, dioxins do not affect the viability of primary cultured placental trophoblast cells in vitro. 23 There are, however, differences in the reported doses of TCDD required to induce cell apoptosis. Finally, TCDD exposure increases early fetal loss, possibly due to an endocrine imbalance that leads to placental insufficiency and compromised embryonic circulation. 24
In the present study, 0.1-10 nM of TCDD, equivalent to a high human exposure level,21,25 did not affect the functional behavior, differentiation or protein expression of EVT-derived cell lines with respect to the HIF1A-VEGF system. This is consistent with what has been observed in the Japanese Yusho study, which followed pregnant women exposed to dioxins. In these women, despite an increased incidence of fetal growth restriction and miscarriage, there was no increase in the incidence of severe preeclampsia, the expected consequence of trophoblast dysfunction.25–27
TCDD and HxCDD inhibit growth by decreasing insulin-like growth factor 1 (IGF-1) signaling in rats. 28 Both TCDD and PCDD/PCDF also inhibit hCG secretion in human placental JEG-3 cell line. 21 These observations taken together with our results suggest that the effect on fetal growth may be the result of an endocrine mechanism rather than impaired trophoblast function.
Footnotes
Acknowledgement
The authors thank A. Noguchi for her technical assistance.
This work was supported in part by a grant-in-aid from the Japan Ministry of Education (23591596) and the Ministry of the Environment, Japan (C-0903).