
Abstract
The majority of breast cancer patients are resistant to chemotherapy or radiotherapy due to the down-regulation or lack of caspase-3 expression. Capsaicin was found to inhibit cancer cell growth in caspase-3-deficient human breast cancer cells. This study aimed to investigate the growth-inhibitive effect of capsaicin and its mechanisms in human breast cancer cell lines, MCF-7 and BT-20. The results showed that cell viability decreased in a dose-dependent manner in both the caspase-3-deficient and non-deficient cells through inducing cell apoptosis and arresting the cell cycle in the S phase. Capsaicin significantly decreased mitochondria membrane potential, induced the cleavage of PARP-1, and decreased procaspase-7 expression in both cells. Apoptosis-inducing factor (AIF) was distinctly released from mitochondria and translocated into the cytoplasm and nucleus in MCF-7 cells (52.9%), but not in BT-20 cells (2%) after treatment with 200 μM of capsaicin for 24 hours. Capsaicin inhibited breast cancer cell growth through inducing cell apoptosis and cell cycle arrest in the S phase. This apoptotic effect could be induced through the mitochondrial pathway, and PARP-1 subsequently cleaved by activation of caspase-7. The application of capsaicin in clinical therapy could be useful for breast cancer patients.
Keywords
Introduction
Breast cancer is one of the most malignant neoplasms worldwide1,2 and is the fourth leading cause of cancer death among Taiwanese in Taiwan. 3 The elimination of breast cancer cells through the induction of breast cancer cell apoptosis plays an important role in breast cancer therapy.4,5 The activation of the caspase family, especially caspase-3, is a vital feature in apoptotic processes.6,7 Also, it has been demonstrated that caspase-3-deficient human breast cancer cells, such as MCF-7 cells, are resistant to apoptotic stimuli and are defective in response to chemotherapy and radiotherapy.7–9 Unfortunately, about 75% of breast cancer tissues are down-regulated by or lack caspase-3 expression; 10 this is considered to be associated with the resistance to chemotherapy and radiotherapy in breast cancer patients.8,9 Therefore, developing an additional therapeutic strategy and clarifying the mechanism of treatment of breast cancer through the caspase-3 independent pathway are strongly recommended.
Capsaicin (8-methyl-N-vanillyl-6-nonenamide), a major pungent ingredient in red peppers of the genus capsicum, has been demonstrated to inhibit carcinogenesis through the induction of apoptosis in human tumor cells, such as colon cancer, 11 hepatoma, 12 breast cancer, 13 prostate tumor 14 and leukemic cells. 15 Our previous study found that capsaicin induced the apoptosis of MCF-7 cells, a caspase-3-deficient human breast cancer cell, in a dose-dependent manner, and that the molecular mechanism of capsaicin-induced apoptosis was through the caspase 3-independent pathway. 13 We suggested that capsaicin could be effective in inhibiting the growth of breast cancer cells, including those that are down-regulated by caspase-3, and that clarifying the mechanism of the action of capsaicin on the induction of apoptosis in human breast cancer cells is important for therapeutic application.
In this study, we investigated the growth-inhibitive effect of capsaicin both in BT-20 cells, non-caspase-3-deficient human mammary adenocarcinoma cells and in MCF-7 cells, caspase-3-deficient human breast cancer cells. Moreover, we sought to understand the mechanisms of capsaicin-induced apoptosis through the estimation of changes in cell viability, cell cycle distributions, mitochondrial membrane potential (MMP), poly (ADP-ribose) polymerase-1 (PARP-1) cleavage, procaspase-7 activation and the translocation of apoptosis-inducing factor (AIF) in both breast cancer cells.
Materials and methods
Cell culture
The human breast cancer cells, MCF-7 and BT-20, were purchased from American Type Culture Collection (Manassas, Virginia). Cells were maintained at 37°C in 5% CO2 and grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 IU/mL penicillin and 100 mg/mL streptomycin (Invitrogen, Carlsbad, California, USA).
Cell viability assay
Cell viability was evaluated by the 4-[3-(4-Iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzenedisulfonate (WST-1) assay (Bio Vision, Mountain View, California, USA). 16 The WST-1 assay measured the activity of mitochondrial dehydrogenases in viable cells by reducing tetrazolium salt to formazan. Briefly, cells (2 × 104/well) were seeded in 96-well plates overnight, followed by capsaicin treatment for 3 days. Then, WST-1 was added to each well and the cells were incubated at 37°C for 4 hours. Finally, plates were measured at OD450 and referenced at OD620 with a microplate reader (SunriseTM, Tecan Groug LTD, Austria). At least three wells per group were analyzed and repeated three times.
Cell cycle analysis
The cell cycle was determined by propidium iodide (PI) staining. 17 Briefly, cells were fixed with 70% ethanol overnight at -20°C and washed with PBS twice by centrifugation (500 × g, 10 minutes, 4°C). Then, the cells were stained with 20 μg/mL PI, 0.1% Triton X-100 and 20 μg/mL RNase A for 1 hour at room temperature. Finally, 2 × 104 cells were analyzed by flow cytometry and CXP analysis software.
Antibodies and reagents
Capsaicin and monoclonal mouse anti-β-actin antibody were purchased from Sigma (St. Louis, Missouri, USA). Polyclonal rabbit anti-PARP-1 and anti-procaspase-7 antibodies were obtained from Cell Signaling Technology (Beverly, Massachusetts, USA). Anti-TRVP1 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, California) and anti-AIF antibody was a gift from Professor KC Chow. HRP-conjugated goat anti-mouse and mouse anti-rabbit IgG antibodies were obtained from Amersham (Freiburg, Germany). Rhodamine-conjugated rabbit anti-mouse was obtained from Jackson Laboratory (West Grove, Wisconsin, USA). Other materials and reagents not specified were obtained from Sigma or Merck.
Western Blot
This technology was based on Burnette’s protocol, 18 and had some modification. Briefly, cells were harvested in lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM glycerolphosphate and 1 mM Na3VO4) with a protease inhibitor mixture (1 mM phyenylmethylsulfonyl fluoride, 10 mg/mL leupeptin and 10 mg/mL aprotinin). Protein concentrations in lysate were determined by Bradford assay 19 using Commassie brilliant blue G-250. Calibration curve was prepared with bovine serum albumin. The lysate was heated at 95°C for 5 minutes in a sample buffer. Fifty micrograms of protein were separated by 8% polyacrylamide gels and transferred to PVDF membranes (Millipore, Bedford, Massachusetts, USA). The membranes were blocked for 1 hour with 5% non-fat dried milk in PBST at room temperature, and then were incubated with specific primary antibody overnight at 4°C. Later, membranes were washed three times with PBST and incubated with HRP-conjugated secondary antibody for 1 hour at room temperature. After washing with PBST three times, the protein signals were detected by an enhanced chemiluminescence detection system (Amersham Biosciences, Piscataway, New Jersey, USA).
Mitochondrial membrane potential detection
MMP was detected by 3,3'-dihexyloxacarbocyanine (DiOC6). 20 Briefly, cells were incubated with 40 nM DiOC6 (Calbiochem, San Diego, California, USA) for 15 minutes at 37°C after capsaicin treatment. Then, 2 × 104 cells were collected and subsequently analyzed by flow cytometry (Beckman/Coulter, Miami, Florida, USA), with a 488-nm excitation wavelength and a 525-nm emission wavelength. Finally, the data were analyzed by CXP analysis software.
Confocal microscopy analysis
This analysis was performed as previously described. 21 Simply, cells were grown on 12-mm glass coverslips overnight, and then were treated with capsaicin for 24 hours. The cells were fixed in 4% paraformaldehyde in PBS for 10 minutes, and then permeabilized with 0.1% Triton X-100 in PBS for 10 minutes at room temperature. After washing with PBS three times, cells were blocked with 5% goat serum in PBS for 30 minutes and incubated with primary antibody in 5% goat serum overnight at 4°C. Then, cells were washed with PBS and incubated with secondary antibody for 2 hours at room temperature, and then washed with PBS and stained nucleus with 4′,6-diamidino-2-phenylindole (DAPI), a fluorescent stain that binds strongly to DNA. Finally, the cells were analyzed with a confocal microscope (LSM510, Zeiss, Chiacago, Illinois, USA). The fluorochrome was excited with an argon laser and UV light of appropriate wavelength. Images were captured with Zeiss LSM 510 software in 1-μm sections and exported to the image processing software, Adobe Photoshop (Adobe Systems, San Jose, California, USA) for further analysis.
Statistical analysis
Experimental results are presented as the mean ± SD. A Mann-Whitney U test was used to estimate the difference between two groups. A one-way ANOVA test was used to detect the difference in cell viability among three, then a Scheffe correction was performed to check statistically significant differences between groups. A p value of less than 0.05 was considered significant. The data were analyzed using SPSS version 17.0 (SPSS Inc., Chicago, Illinois, USA) statistical software.
Results
In order to examine whether BT-20 and MCF-7 breast cancer cells were qualified for our experimental model, the expressions of TRPV1, the receptor for capsaicin, were demonstrated in both the BT-20 and MCF-7 breast cancer cells (Figure 1).
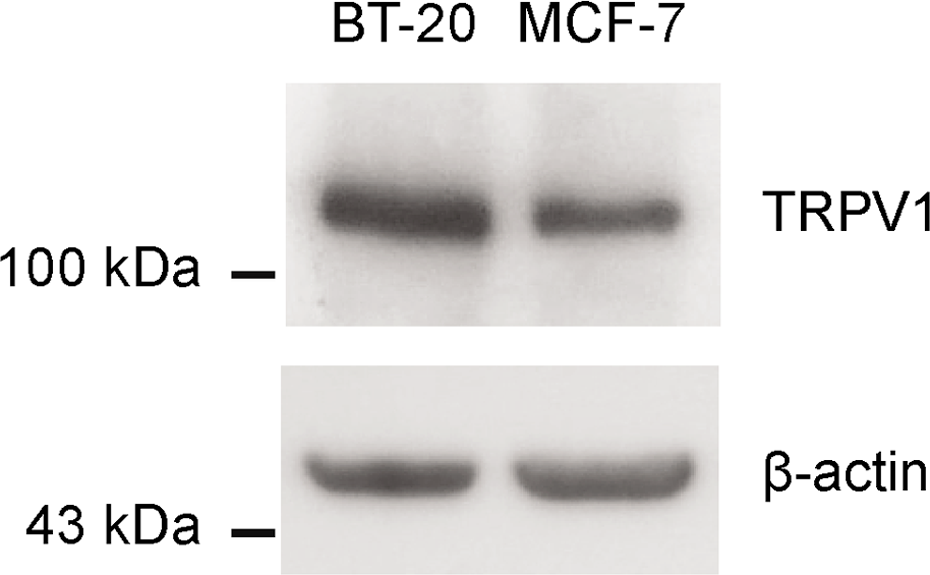
The expression of TRPV1 in breast cancer cells. To evaluate the expression of TRPV1, Western blot analysis was performed.
The survival rate was estimated to predict the effective concentration of capsaicin. The cell survival rate was decreased in a dose-dependent manner in both the BT-20 and MCF-7 cells when they were treated with 100 μM of capsaicin (Figure 2). Moreover, we found that the survival rates were 36.24% ± 2.3% and 58.50% ± 2.5% for BT-20 and MCF-7 cells, respectively, when both cells were treated with 200 μM of capsaicin for 72 hours. We hypothesized that the 200 μM of capsaicin could have a potentially effective concentration of EC40 and EC60 for MCF-7 and BT-20 cells, respectively, to decrease cell survival, and that there was a non-significant difference in the growth-inhibitory effect when treating with 200 μM to 400 μM of capsaicin in both cells; we used the dose of 200 μM of capsaicin for our subsequent investigation.

The effect of capsaicin on cell viability of breast cancer cells. Cells were treated with 50, 100, 200, 300, 400 and 500 μM capsaicin, and 0.05% DMSO (control), respectively, for 72 hours. The cell viability was analyzed using a WST-1 assay. A one-way ANOVA test was used to detect the difference in cell viability among eight groups and a Scheffe correction was performed to check statistically significant differences between groups. ** represents the p value <0.0001.
The cell cycle distributions between untreated and treated cells are shown in Figure 3A. After treatment with 200 μM of capsaicin for 72 hours, there was a significant increase in apoptotic cells, distributed in the sub-G1 phase, in treated compared to untreated BT-20 cells (p = 0.01) and MCF-7 cells (p = 0.02; Figure 3B). The cell cycle distributions were also significantly arrested in the S phase in both cells after treatment with 200 μM of capsaicin for 72 hours (Figure 3C).

The effect of capsaicin on the cell cycle of breast cancer cells. (A) The cell cycle distributions of untreated and treated cells. (B) Significantly increased apoptotic cells, distributed in the sub-G1 phase, in treated and untreated BT-20 cells (p = 0.01) and MCF-7 cells (p = 0.02) after treatment with 200 μM of capsaicin for 72 hours. (C) The comparison of cell cycle distributions of untreated and treated cells. A p value of less than 0.05 between the control group and each capsaicin-treated group was considered significant.
The percentages of cells revealing a decrease of MMP after treatment with 200 μM of capsaicin for 1, 2, 4, 8, and 24 hours, are shown in Figure 4. A significantly increased percentage of cells with a decrease in MMP was found in both BT cells (p = 0.01) and MCF-7 cells (p = 0.003) after they were treated with 200 μM of capsaicin for 24 hours, compared to the untreated cells (control).
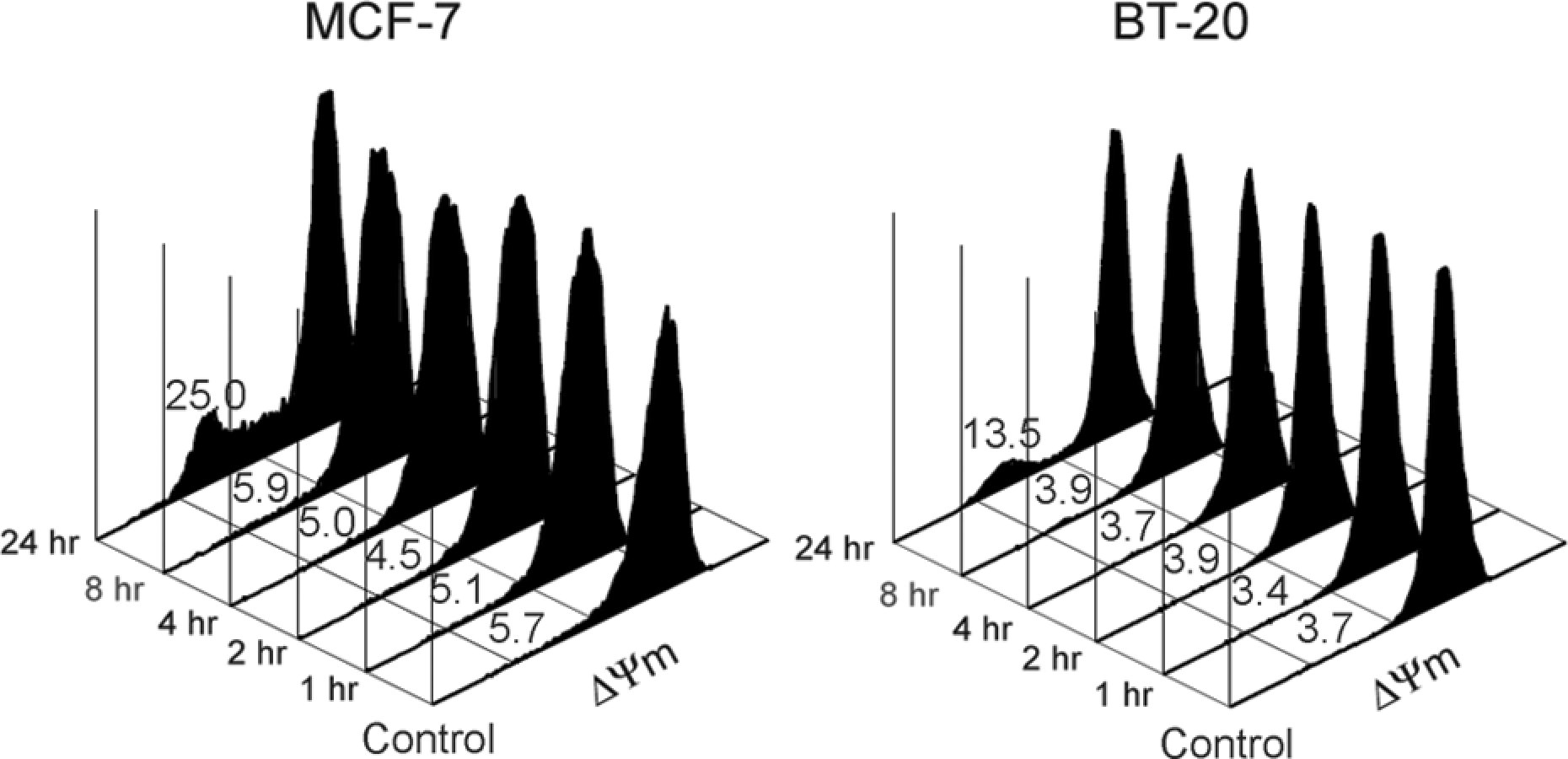
The percentage of cells exhibiting a decrease of mitochondria membrane potential (ΔΨm) under capsaicin treatment. Cells were treated with 200 μM capsaicin for 1, 2, 4, 8 and 24 hours, respectively. After 3,3'-dihexyloxacarbocyanine (DiOC6) staining, the mitochondria membrane potential of capsaicin-treated cells was analyzed by flow cytometry.
The cleavages of PARP-1 after cells were treated with capsaicin at increasing doses for 24 hours, as well as after treatment with 200 μM of capsaicin for different lengths of time are shown in Figure 5. Capsaicin induced the cleavage of PARP-1 in a dose-dependent manner in both groups of breast cancer cells (Figure 5A). Moreover, an induction of the cleavage of PARP-1 in an obviously time-dependent manner after treatment with 200 μM of capsaicin for 4 hours was found in MCF-7 cells, but not in BT-20 cells (Figure 5B). In addition, the results of Western blot demonstrated that procaspase-7 was decreased in both MCF-7 and BT-20 breast cancer cells in a time-dependent manner when they were treated with 200 μM of capsaicin, particularly for MCF-7 cells (Figure 6).

The cleavage of PARP-1 after capsaicin treatment. (A) Cells were treated with increasing doses of capsaicin for 24 hours. (B) Cells were treated with 200 μM of capsaicin for different lengths of time. Equal amounts of whole cell lysates were separated by 8% SDS-PAGE, and PARP was detected by Western blot analysis. β-actin was used as a control for equal loading of protein samples.
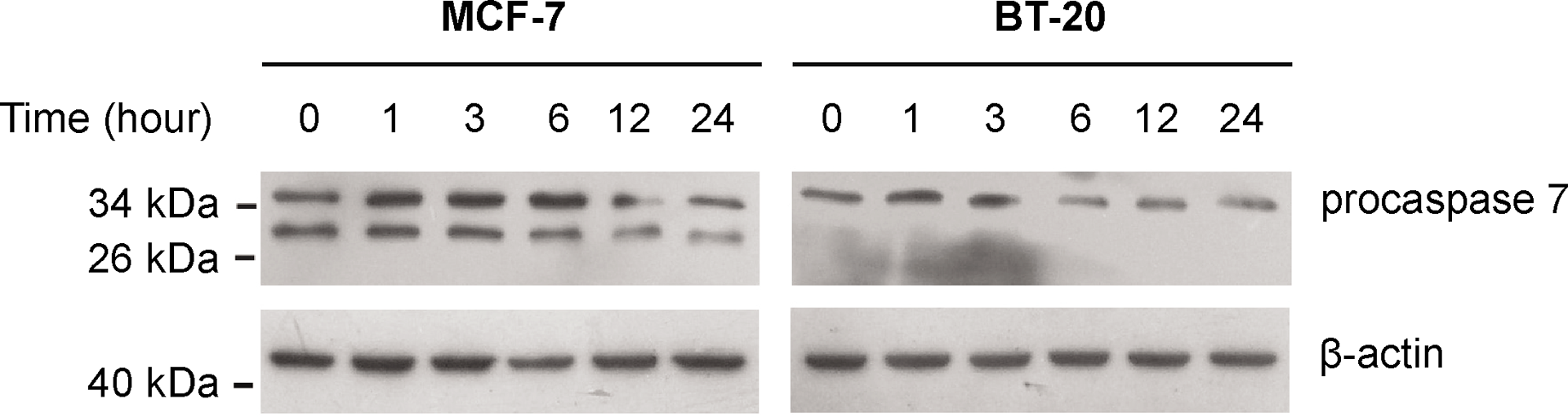
The effect of capsaicin on the activation of procaspase-7 in breast cancer cells. Both breast cancer cells were treated with 200 μM of capsaicin for different lengths of time. Equal amounts of whole cell extracts were separated on 15% SDS-PAGE gels, and procaspase-7 was detected by Western blot analysis.
AIF was distinctly released from mitochondria and translocated into the cytoplasm and nucleus of MCF-7 cells (52.9%), but this was not obvious in BT-20 cells (2%) after both cells were treated with 200 μM of capsaicin for 24 hours (Figure 7). Meanwhile, we found that the mitochondria seemed either shorter or denser in both cells after capsaicin treatment.

Capsaicin induced apoptosis-inducing factor (AIF) translocation in breast cancer cells. The AIF was released from mitochondria and translocated into the cytoplasm and nucleus in MCF-7 cells (left panel, 52.9%) and BT-20 cells (right panel, 2%) after both cells were treated with 200 μM of capsaicin for 24 hours; the translocation of AIF (arrowhead) was detected by confocal microscopy.
Discussion
More and more experimental studies showed that capsaicin can inhibit cell proliferation through induction of apoptosis in various human tumor cells,11–15 while others reported that capsaicin may promote tumor formation. There is controversy that exists between anti-cancer and carcinogenic effects of capsaicin.22,23 This study revealed the cancer-preventing ability of capsaicin in human breast cancer involved by a series of molecular events as shown in results.
Successfully inhibiting the growth of cancer cells using therapeutic strategies is mostly dependent on their ability to trigger tumor cell death.24–26 The activation of apoptosis is mediated mainly by effector caspases, which encompass caspase-3, -6 and -7. 27 Breast cancer patients generally exhibit a reduced sensitivity for anti-cancer therapies; 10 the resistance partially results from the down-regulation of caspase-3 activity, which is the significant executioner caspase, in breast cancer patients.8–10,28
In the present study, we demonstrated that both BT-20 and MCF-7 breast cancer cells revealed the expressions of TRPV1, the receptor for capsaicin. The cell viabilities and survival rates of both cells were significantly decreased in a dose-dependent manner when they were treated with different concentrations of capsaicin, and significantly increased numbers of apoptotic cells and increased induction of cell cycle arrest in the S phase were found in caspase-3-deficient MCF-7 cells and non-caspase-3-deficient BT-20 cells, after they were treated with 200 μM of capsaicin for 72 hours. These data suggested that capsaicin indeed inhibited breast cancer cell growth through inducing cell apoptosis and cell cycle arrest in the S phase, and more importantly, the mechanism could be through a caspase 3-independent pathway, so the application of capsaicin in clinical therapy could be useful for breast cancer patients.
Two distinct pathways, the death receptor pathway (extrinsic pathway) and the mitochondrial pathway (intrinsic pathway), converge on caspase activation.29,30 The extrinsic pathway involves the activation of death receptor, triggered by a death-inducing signal, which in turn activates procaspase 8 and 3, and subsequently induces apoptosis. The intrinsic pathway is mediated by two pro-apoptotic members of the Bax and Bak family. The dimeric pro-apoptotic proteins are inserted into the mitochondrial membrane, and form channels through which cytochrome c escapes into the cytosol. 30 Then, cytochrome c, together with some cofactors, activates caspase-9. The activated caspased-9 leads to activation of one of the effector caspases, caspase 3 or 7, which cleaves an inhibitor of caspase-activated DNAase and produces PARP, resulting in ~180-bp DNA fragmentation.29,31 It has been demonstrated that caspase-7 is highly related to caspase-3, and both caspases have common substrates and could potentially compensate for each other. 32 In our previously published study, we found that increased expression of p53 and Bax and decreased expression of Bcl-2 were found after MCF-7 cells were treated with 150 μM of capsaicin for 24 hours. 13 We therefore hypothesized that the mechanism of capsaicin-induced breast cancer cell apoptosis could be through activation of mitochondrial and caspase-7 pathways. In the present study, we demonstrated that a significantly decreased MMP was found in both breast cancer cells after they were treated with 200 μM of capsaicin for 24 hours. These data indicated that capsaicin could directly decrease MMP and cause mitochondrial dysfunction. Moreover, we found that capsaicin induced the cleavage of PARP-1, not only in BT-20 cells, but also in caspase-3-deficient MCF-7 cells, and that procaspase-7 was decreased in both cells in a time-dependent manner, particularly MCF-7 cells, after treatment with 200 μM of capsaicin. We suggested that capsaicin-induced mitochondrial dysfunction could activate an intrinsic pathway and result in the cleavage of PARP-1 through a caspase 7-dependent pathway.
AIF is a phylogenetically-conserved mitochondrial inter-membrane flavoprotein which is required for inducing apoptosis in the mitochondria-mediated caspase-independent apoptosis pathway.33,34 DNA damage-induced PARP-1 activation is an important requirement to initiate a nuclear signal, which communicates to the mitochondria and triggers the translocation of apoptosis-inducing factor from the mitochondria to the nucleus, inducing nuclear chromatin condensation and DNA fragmentation, which results in a caspase-independent pathway of apoptosis.33,34 In this study, we found that the mitochondria seemed either shorter or denser in both cells and that the AIF was distinctly released from the mitochondria into the cytoplasm and nucleus in MCF-7 cells (52.9%), but this was not obvious in BT-20 cells (2%) after both cells were treated with 200 μM of capsaicin for 24 hours. Joseph et al. demonstrated that radio- or chemotherapy-resistant carcinoma cells were more susceptible to AIF-mediated caspase-independent cell apoptosis. 35 We hypothesized that capsaicin would contribute to mitochondrial dysfunction in both non-caspase-3-deficient BT-20 cells and caspase-3-deficient MCF-7 cells; however, the AIF-mediated caspase-independent pathway could be more effective for caspase-3-deficient cells but not for non-caspase-3-deficient cells.
Capsaicin inhibited breast cancer cell growth through inducing cell apoptosis and cell cycle arrest in the S phase. This apoptotic effect could be induced through the mitochondrial pathway, activated by capsaicin-induced mitochondrial dysfunction, and subsequently cleave PARP-1 by activation of caspase-7. Moreover, capsaicin obviously induced AIF-mediated caspase-independent apoptosis in caspase-3-deficient MCF-7 cells. We suggest that the application of capsaicin in clinical therapy could be useful for breast cancer patients.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.