
Abstract
Secalonic acid D (SAD), a cleft palate-inducing teratogen, has been shown to inhibit proliferation/cell cycle progression in association with alteration in the levels of cell cycle regulators, p21 and cyclin E. These studies were conducted to test the hypotheses that p21 and cyclin E play an important functional role in normal human embryonic palatal mesenchymal (HEPM) cell cycle and that their up- and down-regulation, respectively, by SAD is functionally significant to its cell cycle block. Using small interfering RNA (siRNA) to silence p21gene and transient transfection to overexpress cyclin E in control & SAD-treated HEPM cells, cell proliferation was assessed using a combination of cell numbers, thymidine uptake, CDK2 activity and Ki-67 expression. The results showed that silencing of p21 gene, although increased cell proliferation/numbers and CDK2 activity in normal HEPM cells, failed to counteract SAD-induced anti-proliferative effect despite inducing partial recovery of CDK2 activity. Similar effects were apparent with cyclin E overexpression. It is concluded that p21 and cyclin E are important for normal HEPM cell proliferation. However, SAD-induced deregulation of either protein, singly, may not be sufficient to induce anti-proliferative effect. Involvement of other cell cycle proteins such as cyclin D1 or of multiple proteins in SAD-induced cell cycle block needs to be examined.
Keywords
Introduction
Progression of cell cycle and thus cell proliferation is critical for normal development of the secondary palate, as for all other organs in the developing embryo. Impeded or blocked cell cycle progression would lead to reduced cell numbers/density and formation of smaller palatal shelves that fail to make contact. Smaller palatal shelves are a consistent feature of cleft palate induction by many developmental toxicants including the mycotoxin secalonic acid D 1 (SAD), glucocorticoids 2 and retinoic acid. 3
Cell cycle progression is regulated by many proteins both positively and negatively. Cyclin dependent kinases (CDKs) and their cyclin partners are the major positive regulators that promote cell cycle progression. 4 The activation of CDKs, specific for each phase of the cell cycle, is a two-step process that includes the binding of cyclins to their respective CDK partners followed by CDK phosphorylation by the CDK-activating kinase (CAK). The cyclins and CDKs important in the G1 phase include cyclin D and CDKs 4 and 6. Activated CDK4 and 6 phosphorylate the members of the tumor suppressor family Rb (retinoblastoma) and reduce their suppressive influence on the transcription factor E2F. This sequence of events leads to E2F-mediated transcription of genes, including that of cyclin E, necessary for the transition of the cell into the S phase of the cell cycle. 4 Towards the end of the G1 phase, activated cyclin E/CDK2 complex further contributes to Rb phosphorylation and continued transcription of genes essential throughout the S phase. The activity of CDK2 is also essential for the transition of the cells into the G2 phase 5 and for the restriction of DNA replication to a single cycle. 6 The activity of CDK 1 is important in the G2 and M phases. 4 The members of the Cip/kip family (p21, 27 and p57) and of the INK4 family (p15, 16, 18 and 19) of proteins constitute the major negative cell cycle regulators active either against a wide range of CDKs and other kinases or specifically against G1 CDKs, respectively. 4,5 p53 and other check-point proteins also negatively regulate the cell cycle. Xenobiotics can either promote or hinder the progression of cell cycle by affecting the expression and/or the activity of one or more of these regulators.
Secalonic acid D (SAD) is a mycotoxin produced by the fungus Pencillium oxalicum in grain commodities, 7 with a reported 80% of grain dust samples being positive for its presence. Its potential for adverse human health effects, especially during embryonic development, should be considered significant based on its common presence as a contaminant in corn in the Unites States of America 7 and elsewhere 8 and its capability to induce cleft palate (CP) in animals. 1 Cleft palate induced by SAD in mouse embryos is associated with smaller palatal shelves that fail to elevate and make pre-fusion contact. 1,9 Subsequent studies in our laboratory using mouse and human embryonic palatal mesenchymal (MEPM, HEPM) cells as well as mouse embryonic palatal shelves in vivo 10,11 have shown a reduction in palatal mesenchymal cell numbers, resulting from reduced cell proliferation. However, neither a reduction in extracellular matrix components nor an increase in cell death contributed to shelf size reduction. In HEPM cells, it was further shown that SAD blocks the cell cycle at the G1/S transition. In both HEPM cells and in gestation day 12 murine embryonic palates, exposure to SAD resulted in reduced activity of CDK2, a G1/S-specific CDK. Among the mechanisms of CDK2 activation, neither the expression/level of CDK2 itself nor the activity of its activating kinase (CAK or CDK7) were affected. However, the level of its binding partner cyclin E was reduced and the level of p21 protein, a negative regulator, was increased. 12 It is widely accepted that p21 is a downstream component of many signaling (PKA, PKC, PI-3-kinase, tyrosine kinases such as src) pathways 13,14 more than one of which are affected by SAD. 15,16 p21 may also have a direct role in the mediation of the anti-proliferative effects of chemical agents such as beta-sitosterol, 17 natural alkaloid lycorine 18 and others possibly including SAD. Cyclin E also is likely to modulate the proliferative effects of agents acting not only via PKA pathway 16 but also others.
Both an increase in p21 and a decrease in cyclin E level can lead to reduced CDK2 activity and a cell cycle block at G1/S transition. Nevertheless, since the normal function of these genes in HEPM cells has not been investigated, it is difficult to ascribe functional significance to these effects of SAD. Current studies utilized silencing of p21 gene using small interfering RNA (siRNA) and overexpression of cyclin E to test the hypotheses that p21 and cyclin E genes have a significant role in the regulation of CDK2 activity and proliferation of HEPM cells and that deregulation of p21 and cyclin E by SAD is relevant to its anti-proliferative effect on HEPM cells.
Methods
Materials
HEPM cells were obtained from ATCC and were used between passages 10 and 13 in this study. SAD was extracted and purified according to Reddy et al. 19 Antibodies against p21 were obtained from Santa Cruz Biotech (Santa Cruz, California, USA). p21 siRNA kit was obtained from Upstate Technology (Lake Placid, New York). Chemiluminescence reagents were purchased from Bio-Rad (Hercules, California). Cell culture reagents were obtained from Gibco (Gaithersburg, Maryland, USA). All other chemicals used were from Sigma (St. Louis, Missouri, USA). The DNA sequence encoding the full length human cyclin E with EcoR I and Sal I restriction sites at the 5′ and 3′ end were synthesized from the HU4 cyclin E DNA obtained from Dr Baldassare (Washington University, St. Louis, Missouri, USA) by polymerase chain reaction (PCR). The primers 5’-CGGAATTCCCATGAAGGAGGACGGCGGCGCGG-3’ (forward primer) and 5’-GAGTCGACGCGCCATTTCCGGCCCGCTGCTCTGC-3’ (backward primer) were used for the PCR. The PCR product was digested with EcoR I and Sal I and cloned into the bicistronic pAAV-IRES-GFP expression vector (Invitrogen, Carlsband, California, USA) to obtain pAAV-cyclin E-IRES-GFP vector that results in simultaneous expression of both GFP and cyclin E proteins so that cyclin E expression can be gauged by the extent of GFP expression.
Cell cultures
HEPM cells (~50,000) were plated into 35 mm wells (in a six-well plate) and were cultured according to Dhulipala et al. 11 Cells were exposed to SAD at IC50 of 3.8 μg/mL of medium as determined in previous studies 11 or vehicle Dimethyl-sulfoxide (DMSO) 0.1% in the medium) twice at a 24-hour interval after transfection with siRNA sequences (described below). Following 48 hours of exposure, the cell sheet was washed with Phosphate-buffered saline (PBS), dissociated from the plates by incubating with 0.05% trypsin and 0.5% EDTA, neutralized with the medium and, following centrifugation, preserved at –80°C as a pellet for further use (Western analysis) or processed immediately for performing cell counts.
Silencing of p21 gene using siRNA
Preliminary experiments were conducted to determine the amount (20 pM) of siRNA needed to inhibit SAD-induced p21 completely. This amount of p21 siRNA and equivalent amount of control RNA (cRNA) were used in all the experiments described. Transfection was performed according to the manufacturer’s recommendation. Briefly, 5 µL of Lipofectamine 2000 (Invitrogen) and 20 pM of cRNA or p21 siRNA (siRNA) pool (Upstate Biotechnologies, Lake Placid, New York, USA) were diluted separately in 250 µL of serum-free Opti-MEM and incubated at room temperature for <5 min. The diluted cRNA/siRNA pools were then mixed with the diluted lipofectamine 2000, gently mixed and incubated at room temperature for 20 min. These cRNA/siRNA-lipofectamine 2000 complexes were then added on to the cell cultures. The cultures were incubated at 37°C for 6 hours. After the 6-hour transfection period, the medium and the siRNA-lipofectamine 2000 complexes were removed, the wells washed twice with opti-MEM and 2 mL of fresh medium (with 5% fetal calf serum (FCS)) was added. The cultures were then allowed to recover for 48 hours with medium (with 5% FCS) changes every 24 hours. At the end of the 48-hour recovery period, the cells in two wells from each transfection group (cRNA and siRNA) were harvested and cell numbers were evaluated. The remaining wells in each group were treated twice at 24-hour interval with either the vehicle (0.1% DMSO) or with 3.8 µg of SAD/mL (IC50) as described above. The cells in each group were collected, pelleted and used for immunoblot analysis for p21. All the transfection experiments were conducted in medium containing 5% FCS and no antimicrobials.
Cell proliferation
Cell number was estimated using standard counting technique with hemocytometer according to Hanumegowda et al. 10 3H-thymidine uptake was measured according to Dhulipala et al. 12 The amount of 3H-thymidine incorporated in 5% trichloroacetic acid-insoluble fraction was normalized to the protein levels and expressed as dpm/mg of protein.
Immunoblots for p21 and β-actin
Cell extracts were prepared and immunoblots performed as described by Dhulipala et al. 12 Band intensity was measured densitometrically.
Cyclin-dependent kinase 2 activity assays
Effect of SAD, on the cyclin/CDK2 activity was assessed by performing kinase assays according to Baghdassarian et al. 20 Briefly, the control and SAD-treated HEPM cells from cRNA and siRNA groups were washed with PBS and lysed with buffer containing 0.05 M Tris-HCL, pH 8, 0.15 M NaCl, 0.5% NP-40, 10% glycerol, 1 mM DTT, 1 mM EDTA, 25 mM β-glycerophosphate, 5 mM NaF and protease inhibitors (10 μg/mL soyabean trypsin inhibitor, 1 μg/mL leupeptin, 1 μg/mL aprotinin, 75 μg/mL phenylmethylsulfonyl fluoride). The protein concentration was estimated by the method of Bradford. 21 The lysates were pre-cleared by adding pre-immune rabbit serum for 30 mins, followed by two incubations with protein A beads. Lysates (100 μg of protein equivalent) were then incubated with 5 μg of CDK2 antibodies overnight at 4°C on a rocker and the immune complexes were precipitated using protein A beads, washed twice with both lysis buffer as well as kinase buffer. The beads were re-suspended in kinase buffer containing 50 mM HEPES, (pH 7.5), 10 mM MgCl2, 1 mM DTT and 50 μM ATP and 10 μCi of [γ32 P]-ATP. Histone H1 (10 μg) was used as substrate for activity assays. The mixtures were incubated at 37°C for 30 min. Samples were denatured in Laemilli buffer, boiled for 3 min and subjected to 12% SDS-PAGE. The gels were dried and subjected to autoradiography. The intensity of the phosphorylated substrate band was measured densitometrically and considered representative of the CDK2 activity in the samples.
Overexpression of cyclin E
To assess the functional significance of cyclin E in the anti-proliferative effects of SAD, this protein was overexpressed in HEPM cell line following normal transient transfection protocol. The full length cyclin E gene cloned into the pAAV-IRES-GFP (pAAV-IRES-cyclinE-GFP) along with the pAAV-IRES-GFP vector was used in these experiments. Cultures transfected with pAAV-IRES-GFP (without cyclin E) were used as controls. The transfected cells (5%–10%) were identified under UV light by assessing the GFP expression.
To assess the functional significance of cyclin E in palatal cell proliferation and in the anti-proliferative effect of SAD, the HEPM cells were grown on poly-lysine coated coverslips (BD biosciences, San Diego, California, USA) and transfected with the above-mentioned constructs 24 hours later using ~1 µg of DNA for 6 hours using Lipofectamine 2000 according to the manufacturer’s protocol. After a medium change and a 24-hour recovery, the cells were treated with 3.8 µg of SAD/mL or with DMSO (vehicle). The functional significance of cyclin E in the SAD induced anti-proliferative effects was judged by a subjective assessment of the reversal of the anti-proliferative effects of SAD following cyclin E overexpression using nuclear Ki-67 immunostaining as an index.
Immunocytofluorescence
Cells, grown and treated as described above, were fixed in 4% paraformaldehyde for 15 min at 37°C, rinsed in wash buffer (PBS + 0.1 – 0.2% Aurion BSA + 15 mM NaN3), permeabilized in PBS + 0.1% Triton-X 100 for 15 min and incubated in PBS + 5% BSA + 5% normal goat serum for 30 min at 37°C. The cells were then rinsed 3 times in wash buffer followed by incubation with anti-mouse GFP Alexa fluor 488 (1:100, EX-494 nm, EM-517 nm; Molecular Probes, Eugene, Oregon, USA) and Santa Cruz primary rabbit polyclonal antibody for Ki-67 (1:100) overnight at 4°C, washed and incubated with goat anti-rabbit immunoglobulin G (IgG) antibody conjugated with Alexa Fluor 568 (EX-568 nm, EM -600 nm, Molecular Probes) wash buffer + 1% BSA for 2 hours. The cells were washed again, coverslipped and mounted onto glass slides with 2 µL of Molecular Probes prolonged antifade kit and sealed.
Images of the cells were obtained using a 60× water immersion objective (1.2 NA) on an Olympus IX-70 inverted microscope coupled with a Bio-Rad Radiance-2000 (Hercules, California, USA) confocal system. The GFP vectors labeled with anti-GFP Alexa-488-conjugated antibodies were excited at 488 nm and the signals were collected through a 515/30 nm emission filter. All instrument parameters were optimized including the pinhole apertures for each excitation wavelength. Optical sections (10−15) were obtained at 0.7 µ intervals along the z-axis. The stacks were saved as raw PIC format data and 3-D reconstructed using the software, Metamorph (version 6.0, Universal Imaging Corp, Westchester, Pennsylvania, USA). Images for GFP and the proliferation marker Ki-67 were calibrated independently and overlaid. Specificity of secondary antibody was verified in all experiments by the addition of secondary antibody in the absence of the primary antibody, which showed no fluorescent signal (not shown). Approximately 40−50 cells were imaged for each protein. The number of dividing cells was assessed visually (Ki-67 presence & morphology).
Statistics
The cell number, 3H-thymidine uptake and densitometric values from immunoblots obtained from siRNA and/or SAD treatment groups were compared using Student’s t-test where two groups were compared and ANOVA in conjunction with Student Newman Kaul (SNK) test where more than two groups were compared. A p ≤ .05 was considered significant.
Results
P21 siRNA treatment completely blocks SAD-induced p21 elevation in HEPM cells
Treatment of HEPM cells with vehicle alone or with non-specific (control) RNA (cRNA) alone resulted in a low basal expression of p21 protein (Figure 1). Transfection with 5 ng of p21 siRNA completely blocked the basal p21 levels seen in cells treated with cRNA (Figure 1A). This indicated that the siRNA was very effective in silencing p21 expression. The β-actin levels in these treatment groups showed that equal amount of protein was loaded in each lane (Figure 1).

p21 siRNA silenced p21 expression in human embryonic palatal mesenchymal (HEPM) cells. HEPM cells were transfected either with control RNA (cRNA) or p21 siRNA and treated with secalonic acid D (SAD; cRNA + SAD and siRNA + SAD). Whole cell lysates (n = 3) were immunoblotted for p21 as described in methods. Lane A shows that SAD increased the levels of p21 in the cells treated with cRNA but this increase in p21 expression was abolished in the siRNA-transfected cells. Lane B shows equal loading of cell lysates in each lane as assessed by immunoblotting for β-actin protein.
Silencing p21 gene expression increases cell numbers, thymidine uptake and CDK2 activity in HEPM cells
cRNA equal to the amount of siRNA that completely inhibited SAD-induced increase in p21, although caused some anti-proliferative effect of its own compared to untreated cells, allowed survival of sufficient number of cells for the purpose of this investigation. The number of HEPM cells at the end of the culture period was significantly greater following siRNA treatment (Figure 2A), compared to those following treatment with cRNA, indicating that downregulation of p21 leads to increased cell numbers. Similar increases were also noted in the uptake of 3H-thymidine (an indicator of proliferation) and in the activity of CDK2 in the HEPM cells in which the expression of p21 was silenced compared to those transfected with cRNA (Figures 2B and 3).

A, Effect of p21 gene silencing on human embryonic palatal mesenchymal (HEPM) cell numbers. HEPM cells were transfected either with control RNA (CSi) or p21 siRNA (p21Si) or left untransfected (D) and the number of cells in each transfection group were assessed (n = 3) after 96 hours using a hemocytometer as described in methods. Silencing the expression of p21 in HEPM cells allows for a greater increase in cell numbers compared to those transfected with cRNA. * indicates significant difference (p ≤ .05) compared to D. B, Effect of p21 gene silencing on thymidine uptake (dpm/mg protein) in HEPM cells. HEPM cells were transfected either with control siRNA (cSi) or p21 siRNA (p21Si) and the amount of tritiated thymidine incorporated into the cells in each transfection group during the last 4 hours of the 96-hour culture period was assessed (n = 3) as detailed in methods. Silencing p21 expression in HEPM cells increases the amount of tritiated thymidine incorporation compared to cells transfected with cRNA. * indicates significant difference (p ≤ .05) between transfection groups.

Effect of p21 gene silencing on CDK2 activity in human embryonic palatal mesenchymal (HEPM) cells. HEPM cells were transfected either with control RNA (cRNA) or p21 siRNA (siRNA) after a 96-hour culture period, and the level of CDK2 activity in the two transfection groups was assessed by performing activity assays (n = 3) as discussed in methods. Silencing p21 expression in HEPM cells increased the activity of CDK2 compared to cRNA-transfected cells. * indicates significant difference (p ≤ .05) between transfection groups.
Silencing p21 expression blocked SAD-induced p21 upregulation, partially blocked SAD-induced inhibition of CDK2 activity but failed to block SAD-induced reduction in HEPM cell numbers and thymidine uptake
As expected, treatment with SAD dramatically increased p21 protein level in HEPM cells (Figure 1). Exposure to siRNA completely antagonized SAD-induced p21 upregulation (Figure 1). Also, as expected, exposure to SAD reduced the cell number by ~50% in HEPM cells not transfected with any RNA (Figure 4 , DS compared to D). The magnitude (% of control) of the antiproliferative effect of SAD, at the end of the 48 hours of toxicant treatment, in cells transfected with cRNA (CSi) or p21siRNA (p21Si) was similar to the vehicle-treated controls (Figure 4) suggesting that silencing p21 gene failed to antagonize the anti-proliferative effect of SAD (Figure 4, CSi + SAD compared to p21Si + SAD). Comparison of the actual cell counts in the two transfection groups before and after SAD treatment, however, indicated that silencing of p21 gene increased HEPM cell numbers, as expected and shown earlier, at both time-points compared to cRNA-treated cells (Table 1).

Effect (or lack thereof) of p21 gene silencing on secalonic acid D (SAD)-induced reduction in cell numbers. Human embryonic palatal mesenchymal (HEPM) cells were untransfected (D) or transfected with control RNA (cSi) or p21 siRNA (p21Si) and treated with either the vehicle (D, cSi, p21Si) or with IC50 of SAD (DS, cSi + SAD, p21Si + SAD) as described in the methods. Cell numbers in each group (n = 3) are expressed as % of the respective controls. * indicates significant difference (p ≤ .05) from the respective controls.

Abbreviations: SAD: secalonic acid D, cRNA: control RNA, siRNA: small interfering RNA.
a cRNA and siRNA represent HEPM cells transfected with control RNA and p21 siRNA, respectively.
b Values in parenthesis represent cell numbers as a percentage of post-treatment cRNA and siRNA control group averages. Cell number values represent mean ± SD for three replicate experiments.
Silencing the p21 gene expression almost doubled the amount of thymidine uptake compared to the cRNA group in the absence of exposure to SAD suggesting that, indeed, the increase in cell number resulting from p21 silencing is a result of increased cell cycle (S-phase) activity. Silencing of p21, however, failed to prevent SAD-induced decrease in thymidine uptake (Table 2). Therefore, p21 gene silencing failed to prevent SAD-induced reduction in proliferative activity.
Effect of p21 gene silencing on SAD-induced reduction in 3H-thymidine uptake in HEPM cells (dpm/mg protein) a

Abbreviations: SAD: secalonic acid D, cRNA: control RNA, siRNA: small interfering RNA.
a Values represent mean ± SD for three replicate experiments.
b indicates significant difference (p ≤ 0.05) in SAD-treated group from the respective controls.
The activity of CDK2 in equal amounts of immunoprecipitates derived from cell lysates using CDK2 antibodies (assessed based on the amount of substrate phosphorylation) indicated that silencing p21 expression in the absence of SAD-treatment enhanced the kinase activity in the HEPM cells compared to the respective cRNA group (Figure 3). In the presence of SAD, however, siRNA transfection led only to a small but noticeable increase in CDK2 activity compared to cRNA-transfected and SAD-treated cells (Figure 5). However, the CDK activity in this group was significantly less than that in cells treated with cRNA alone indicating that p21 gene silencing failed to completely reverse SAD-induced reduction in CDK2 activity.

Effect of p21 gene silencing on the secalonic acid D (SAD) induced reduction in CDK2 activity. Human embryonic palatal mesenchymal (HEPM) cells were transfected either with control siRNA (cRNA) or p21 siRNA (siRNA) and treated with SAD (cRNA + SAD and siRNA + SAD). Equal amounts of whole cell lysates (n = 2) were immunoprecipitated with CDK2 antibody and the precipitates were analyzed for CDK2 activity as mentioned in the methods. Silencing p21 expression dramatically enhanced the activity of CDK2 compared to the cRNA group and mildly but noticeably reversed SAD-induced reduction in the CDK2 activity.
Overexpression of cyclin E does not prevent the anti-proliferative effect of SAD in HEPM cells
HEPM cells transfected with the empty pAAV-IRES-GFP vector (control vector group) expressed GFP, whereas those transfected with pAAV-IRES-cyclinE-GFP expressed both GFP and cyclin E at very high levels (Figure 6). Intranuclear presence of the proliferation marker Ki-67, as assessed by immunofluorescence, was detected as dense foci in most cells in both of these vector groups suggesting that most HEPM cells were cycling. Such focal accumulation is known to occur in early G1 phase and represent the reforming nucleoli. 22 Exposure to SAD disrupted the nuclear condensation of Ki-67 and, instead, promoted its perinuclear distribution. Overexpression of cyclin E failed to antagonize both SAD-induced disruption of nuclear Ki-67 condensation and its perinuclear accumulation compared to cells in the pAAV-IRES-cyclinE-GFP vector group (Figure 6). Subjective evaluation of the small number of cyclin E-overexpressing cells also indicated that these cells were multiplying more rapidly than controls and more clearly than the SAD-treated cells using both Ki-67 expression and the number of dividing and separating cells (represented in Figure 6, row 3 compared to row 1 and more clearly to rows 2 and 4) as indices.
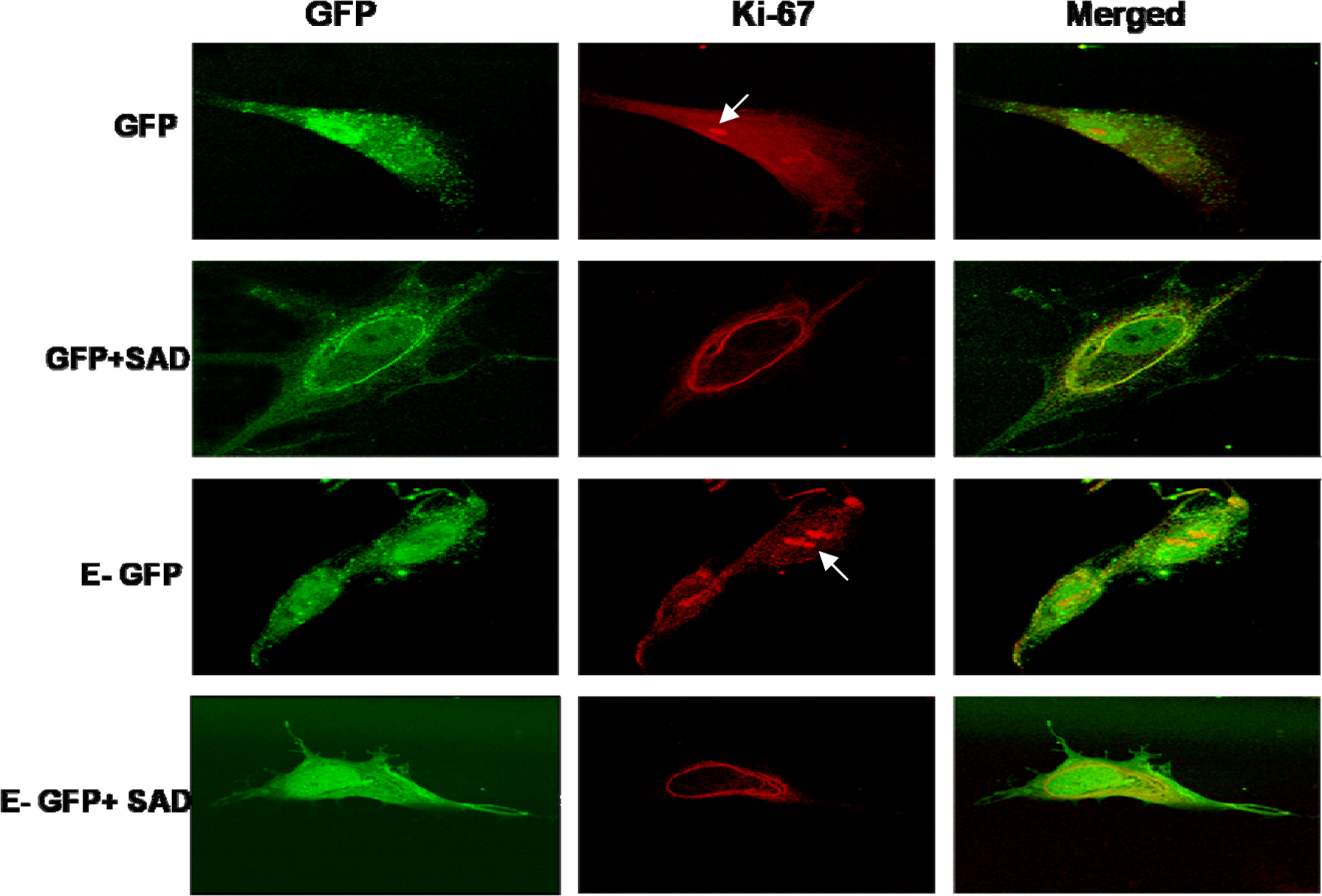
The effect of overexpression of cyclin E on secalonic acid D (SAD)-induced reduction in cell proliferation in human embryonic palatal mesenchymal (HEPM) cells. HEPM cells were transfected with either pAAV-IRES-GFP (GFP) vector or pAAV-cyclin E-IRES-GFP (E-GFP) and treated with SAD (GFP + SAD and E-GFP + SAD). The nuclear localization of the proliferation marker Ki-67 in each treatment group was assessed by immunofluorescence as described in the methods. The intensity of nuclear Ki-67 was used as an index of proliferation. The presence of nuclear Ki-67 in the GFP and E-GFP cells is indicated by the arrows. Ki-67 was not present in the nuclei of the SAD-treated cells transfected with either of the plasmids. The results indicated that, by this subjective assessment, overexpression of cyclin E although appeared to increase nuclear Ki-67 compared to those transfected with control vector did not reverse the anti-proliferative effects of SAD.
Discussion
Sequential activation of cyclin/CDK complexes is essential for the transition of the proliferating cells from the G1 phase into the S phase. 5 Cyclin D1 is known as one of the first cyclins expressed in the proliferating cells and is known to bind to and activate CDK4/6. 23 Cyclin E, together with the Cip/Kip proteins p21 and/or p27, is involved in the regulation of the activity of CDK2. Upon activation, these kinases phosphorylate and inactivate the tumor suppressor protein, Rb, which in turn results in the release and activation of the transcription factor E2F. E2F binds to its response element in and activates the transcription of many genes involved in DNA replication and cell progression through S-phase. 23
In many cell lines, cyclin E enhances while p21 reduces the activity of CDK2. 23,24 The results from our p21 gene silencing and cyclin E overexpression experiments showed, for the first time, that reducing p21 levels or increasing cyclin E expression in cultured HEPM cells would indeed promote cell cycle progression. These results clearly affirm the negative and positive regulatory roles for p21 and cyclin E genes, respectively, in HEPM cell cycle, similar to their roles in many other cell types.
As shown in earlier studies with HEPM cells in vitro as well as the murine embryonic palates in vivo, 12 the results of this study also showed that the CP-inducing mycotoxin, SAD, increases the levels of p21, inhibits the activity of CDK2, impedes with S-phase progression (thymidine uptake) and reduces the cell number in HEPM cell cultures. Similar effects were reported for fibroblast growth factor (FGF) in chondrocytes, 25 for cudraflavone in rat aortic smooth muscle cells, 26 for benzo[a]pyrene in human embryo lung fibroblasts 27 and apicidin in Ishikawa human endometrial cancer cells, 28 among many others. Upregulation of p21 levels in HEPM cells by SAD strongly suggests that the p21 gene silencing should abrogate the anti-proliferative effects of SAD. Whether a failure of p21 silencing in antagonizing the effects of SAD in the present studies reflects a lack of a mechanistic role for p21 gene in the anti-proliferative effects of SAD or a role for other cell cycle genes either alone or in concert with p21 and/or cyclin E needs to be assessed further. The fact that complete p21 silencing produced only a partial reversal of SAD-induced CDK2 inhibition suggests the likelihood of the latter possibility and a partial role for p21. The fact that the deletion or silencing of p21 only partially antagonized the anti-proliferative effects of FGF whereas the effects of benzo[a]pyrene were unaffected in other studies 25,27 not only indicate that the results obtained in our studies are not unique but also supports the contention that multiple players coordinate cellular proliferative response. Whether a similar effect is likely with cyclin E overexpression and whether a combined p21 and cyclin E gene modulation would produce a complete reversal are questions that can be better addressed using cells in which with one or both gene modulations can be made in 100% of cells. The interesting phenomenon of perinuclear accumulation of Ki-67 following SAD-exposure as seen in our studies has not been previously reported. Whether this represents inhibition of nuclear transport of Ki-67 due to the loss of carboxy terminal nuclear targeting signal 22 or other mechanisms needs to be addressed as well. As both p21 and cyclin E are known to act on CDK2 in opposite directions, such an assessment is both relevant and justified. Such a possibility is supported by the findings of Wolfraim et al. 29 showing that a single gene deletion for either p21Cip1 or p27Kip1 had no effect on the ability of TGF-beta to induce G1 arrest in lymphocytes whereas their combined deletion acted in a synergistic manner to impair the response to TGF-beta as assessed by tritiated thymidine incorporation. Another possibility is a rescue of SAD-induced loss of cyclin E or gain of p21 function by compensatory changes in other cell cycle modulators (cyclins D, B, A and/or p27, p57 and/or INK4). Such a mechanism was shown to be operative in p21-mediated MyoD-induced growth arrest of myoblasts. Loss of p21 failed to overcome myoD-induced growth inhibition due to compensatory elevation of another member of the Cip/Kip family of proteins, the p57. 30,31 Whether a reduction of all four INK4 proteins in HEPM cells by SAD 12 indicates compensation for increased p21 levels is unknown.
Cip/Kip family of CDK inhibitors, of which p21 is a member, regulate cell cycle negatively by their obligatory binding to preformed cyclin/CDK complexes. 32 In quiescent cells, low basal level of cyclinE/CDK2 complex is kept inactive by the Cip/Kip inhibitors present. Mitotic stimulation leads to an abundance of the cyclin D/CDK4/6 complex that binds to and is activated by the Cip/Kip inhibitors. The active cyclin D/CDK 4/6 will phosphorylate Rb and initiate the progression of cell cycle as well as the transcription of downstream cyclins including cylin E. The newly formed cyclinE/CDK2 complex, being free from inhibition by the Cip/Kip inhibitors because of their sequestration within the cyclinD/CDK4/6 complex, is then activated leading to the hyperphosphorylation of Rb and progression of the cell through the G1/S transition. In cycling cells, inhibition of cell cycle progression by Cip/Kip inhibitors such as p21 will require an increase in their expression large enough to saturate the cyclin D/CDK4/6 sequestration and to overcome the threshold for cyclin E/CDK2 binding. In SAD-exposed cells, however, since the levels of cyclin D, CDK4, cyclin E and CDK2 are all reduced, 12 it is possible that the amounts of the preformed cyclin D/CDK4 and cyclin E/CDK2 complex and thus their activities failed to reach the threshold needed for the cell progression into the S phase. Under these conditions, the cell-cycle-blocking effect of SAD-induced reduction in the levels/activity of cyclin D/CDK4 and cyclinE/CDK2 complexes themselves may take precedence over that of SAD-induced increase in p21 level. This possibility needs to be examined in future studies. Further, Keenan et al. 33 showed that exogenous cyclin E expression and CDK2 activation in the absence of CDK4 activity can still lead to Rb phosphorylation, E2F release and cell cycle progression in hamster fibroblasts. If these results hold true for HEPM cells, the restoration of cyclin E/CDK2 complex alone in SAD-treated HEPM cells is likely to restore their proliferation even when cyclinD/CDK4 levels are down-regulated by SAD.
Exposure to SAD leads to a reduction in cyclin D1 expression either directly or via its inhibitory effect on murine palatal epidermal growth factor (EGF) levels 34 and/or on protein kinase C 15 and protein kinase A 16 signaling pathways, all critical for cyclin D expression. 35,36 This opens the possibility of having multiple protein targets for SAD in inducing its anti-proliferative effects. For example, cyclin D1 could be an important candidate for assessing the functional significance in the SAD-induced CP because it is the only other cell cycle protein shown to be commonly affected by the toxicant in both HEPM and murine embryonic palates. Matsushime et al. 37 have shown that D type cyclins can act as delayed early response genes to various growth factors (mitogens) in many cell types and that the levels of these cyclins are directly proportional to the mitogen levels. In the case of SAD, reduction in cyclin D1 levels can be either a direct effect of SAD or secondary to SAD-induced reduction in EGF levels. Cyclin D1 which binds to CDK4 is essential for Rb phosphorylation. Even though both cyclin D1-CDK4 and cyclin E-CDK2 are essential for Rb phosphorylation and inactivation, it has been shown that overexpression of cyclin D1 leads to a rapid phosphorylation of Rb compared to a delayed effect noticed with the overexpression of cyclin E in the same cells. This makes cyclin D1 an important candidate for assessing its functional significance in the induction of CP by SAD. The fact that SAD exposure reduced the activity of CDK4/6 in HEPM cells 12 supports this contention. Overall, the results of this study indicate that although cyclin E and p21 have important individual roles in HEPM proliferation, cell cycle proteins other than cyclin E and p21 or a simultaneous alteration of more than one cell cycle protein including p21 and cyclin E may be involved in the induction of cell cycle block by SAD.
Footnotes
Acknowledgements
We would like to acknowledge Dr Baldessare of Washington University (St Louis, Missouri) for his kind gift of the cyclin E construct.
These studies were supported by grants from the University of Missouri Research Council, Pi Chapter of Phi Zeta, National Institutes of Health (grant #DEOD11822), and Alternatives Research and Development Foundation.